 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBoosting t-SNE Efficiency for Sequencing Data: Insights from Kernel Selection
Dec 17, 2025



Dimensionality reduction techniques are essential for visualizing and analyzing high-dimensional biological sequencing data. t-distributed Stochastic Neighbor Embedding (t-SNE) is widely used for this purpose, traditionally employing the Gaussian kernel to compute pairwise similarities. However, the Gaussian kernel's lack of data-dependence and computational overhead limit its scalability and effectiveness for categorical biological sequences. Recent work proposed the isolation kernel as an alternative, yet it may not optimally capture sequence similarities. In this study, we comprehensively evaluate nine different kernel functions for t-SNE applied to molecular sequences, using three embedding methods: One-Hot Encoding, Spike2Vec, and minimizers. Through both subjective visualization and objective metrics (including neighborhood preservation scores), we demonstrate that the cosine similarity kernel in general outperforms other kernels, including Gaussian and isolation kernels, achieving superior runtime efficiency and better preservation of pairwise distances in low-dimensional space. We further validate our findings through extensive classification and clustering experiments across six diverse biological datasets (Spike7k, Host, ShortRead, Rabies, Genome, and Breast Cancer), employing multiple machine learning algorithms and evaluation metrics. Our results show that kernel selection significantly impacts not only visualization quality but also downstream analytical tasks, with the cosine similarity kernel providing the most robust performance across different data types and embedding strategies, making it particularly suitable for large-scale biological sequence analysis.
Enhancing Privacy Preservation and Reducing Analysis Time with Federated Transfer Learning in Digital Twins-based Computed Tomography Scan Analysis
Sep 09, 2025The application of Digital Twin (DT) technology and Federated Learning (FL) has great potential to change the field of biomedical image analysis, particularly for Computed Tomography (CT) scans. This paper presents Federated Transfer Learning (FTL) as a new Digital Twin-based CT scan analysis paradigm. FTL uses pre-trained models and knowledge transfer between peer nodes to solve problems such as data privacy, limited computing resources, and data heterogeneity. The proposed framework allows real-time collaboration between cloud servers and Digital Twin-enabled CT scanners while protecting patient identity. We apply the FTL method to a heterogeneous CT scan dataset and assess model performance using convergence time, model accuracy, precision, recall, F1 score, and confusion matrix. It has been shown to perform better than conventional FL and Clustered Federated Learning (CFL) methods with better precision, accuracy, recall, and F1-score. The technique is beneficial in settings where the data is not independently and identically distributed (non-IID), and it offers reliable, efficient, and secure solutions for medical diagnosis. These findings highlight the possibility of using FTL to improve decision-making in digital twin-based CT scan analysis, secure and efficient medical image analysis, promote privacy, and open new possibilities for applying precision medicine and smart healthcare systems.
EfficientNet in Digital Twin-based Cardiac Arrest Prediction and Analysis
Sep 09, 2025Cardiac arrest is one of the biggest global health problems, and early identification and management are key to enhancing the patient's prognosis. In this paper, we propose a novel framework that combines an EfficientNet-based deep learning model with a digital twin system to improve the early detection and analysis of cardiac arrest. We use compound scaling and EfficientNet to learn the features of cardiovascular images. In parallel, the digital twin creates a realistic and individualized cardiovascular system model of the patient based on data received from the Internet of Things (IoT) devices attached to the patient, which can help in the constant assessment of the patient and the impact of possible treatment plans. As shown by our experiments, the proposed system is highly accurate in its prediction abilities and, at the same time, efficient. Combining highly advanced techniques such as deep learning and digital twin (DT) technology presents the possibility of using an active and individual approach to predicting cardiac disease.
Sequence Analysis Using the Bezier Curve
Mar 18, 2025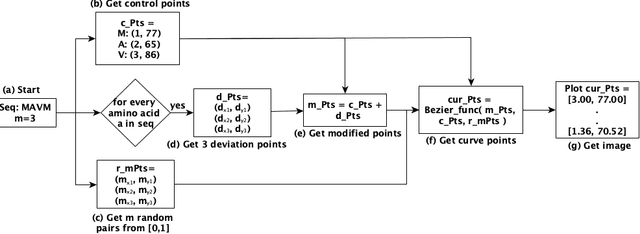

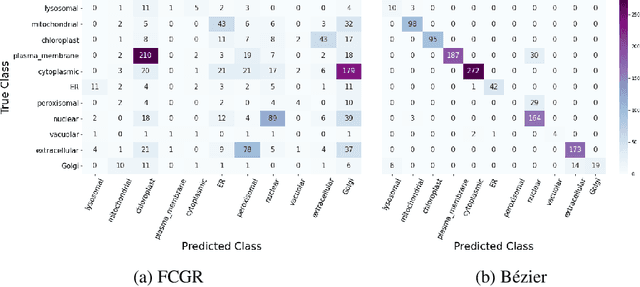
The analysis of sequences (e.g., protein, DNA, and SMILES string) is essential for disease diagnosis, biomaterial engineering, genetic engineering, and drug discovery domains. Conventional analytical methods focus on transforming sequences into numerical representations for applying machine learning/deep learning-based sequence characterization. However, their efficacy is constrained by the intrinsic nature of deep learning (DL) models, which tend to exhibit suboptimal performance when applied to tabular data. An alternative group of methodologies endeavors to convert biological sequences into image forms by applying the concept of Chaos Game Representation (CGR). However, a noteworthy drawback of these methods lies in their tendency to map individual elements of the sequence onto a relatively small subset of designated pixels within the generated image. The resulting sparse image representation may not adequately encapsulate the comprehensive sequence information, potentially resulting in suboptimal predictions. In this study, we introduce a novel approach to transform sequences into images using the B\'ezier curve concept for element mapping. Mapping the elements onto a curve enhances the sequence information representation in the respective images, hence yielding better DL-based classification performance. We employed different sequence datasets to validate our system by using different classification tasks, and the results illustrate that our B\'ezier curve method is able to achieve good performance for all the tasks.
Hilbert Curve Based Molecular Sequence Analysis
Dec 29, 2024Accurate molecular sequence analysis is a key task in the field of bioinformatics. To apply molecular sequence classification algorithms, we first need to generate the appropriate representations of the sequences. Traditional numeric sequence representation techniques are mostly based on sequence alignment that faces limitations in the form of lack of accuracy. Although several alignment-free techniques have also been introduced, their tabular data form results in low performance when used with Deep Learning (DL) models compared to the competitive performance observed in the case of image-based data. To find a solution to this problem and to make Deep Learning (DL) models function to their maximum potential while capturing the important spatial information in the sequence data, we propose a universal Hibert curve-based Chaos Game Representation (CGR) method. This method is a transformative function that involves a novel Alphabetic index mapping technique used in constructing Hilbert curve-based image representation from molecular sequences. Our method can be globally applied to any type of molecular sequence data. The Hilbert curve-based image representations can be used as input to sophisticated vision DL models for sequence classification. The proposed method shows promising results as it outperforms current state-of-the-art methods by achieving a high accuracy of $94.5$\% and an F1 score of $93.9\%$ when tested with the CNN model on the lung cancer dataset. This approach opens up a new horizon for exploring molecular sequence analysis using image classification methods.
Converting Time Series Data to Numeric Representations Using Alphabetic Mapping and k-mer strategy
Dec 29, 2024In the realm of data analysis and bioinformatics, representing time series data in a manner akin to biological sequences offers a novel approach to leverage sequence analysis techniques. Transforming time series signals into molecular sequence-type representations allows us to enhance pattern recognition by applying sophisticated sequence analysis techniques (e.g. $k$-mers based representation) developed in bioinformatics, uncovering hidden patterns and relationships in complex, non-linear time series data. This paper proposes a method to transform time series signals into biological/molecular sequence-type representations using a unique alphabetic mapping technique. By generating 26 ranges corresponding to the 26 letters of the English alphabet, each value within the time series is mapped to a specific character based on its range. This conversion facilitates the application of sequence analysis algorithms, typically used in bioinformatics, to analyze time series data. We demonstrate the effectiveness of this approach by converting real-world time series signals into character sequences and performing sequence classification. The resulting sequences can be utilized for various sequence-based analysis techniques, offering a new perspective on time series data representation and analysis.
Computing Gram Matrix for SMILES Strings using RDKFingerprint and Sinkhorn-Knopp Algorithm
Dec 19, 2024In molecular structure data, SMILES (Simplified Molecular Input Line Entry System) strings are used to analyze molecular structure design. Numerical feature representation of SMILES strings is a challenging task. This work proposes a kernel-based approach for encoding and analyzing molecular structures from SMILES strings. The proposed approach involves computing a kernel matrix using the Sinkhorn-Knopp algorithm while using kernel principal component analysis (PCA) for dimensionality reduction. The resulting low-dimensional embeddings are then used for classification and regression analysis. The kernel matrix is computed by converting the SMILES strings into molecular structures using the Morgan Fingerprint, which computes a fingerprint for each molecule. The distance matrix is computed using the pairwise kernels function. The Sinkhorn-Knopp algorithm is used to compute the final kernel matrix that satisfies the constraints of a probability distribution. This is achieved by iteratively adjusting the kernel matrix until the marginal distributions of the rows and columns match the desired marginal distributions. We provided a comprehensive empirical analysis of the proposed kernel method to evaluate its goodness with greater depth. The suggested method is assessed for drug subcategory prediction (classification task) and solubility AlogPS ``Aqueous solubility and Octanol/Water partition coefficient" (regression task) using the benchmark SMILES string dataset. The outcomes show the proposed method outperforms several baseline methods in terms of supervised analysis and has potential uses in molecular design and drug discovery. Overall, the suggested method is a promising avenue for kernel methods-based molecular structure analysis and design.
EPIC: Enhancing Privacy through Iterative Collaboration
Nov 07, 2024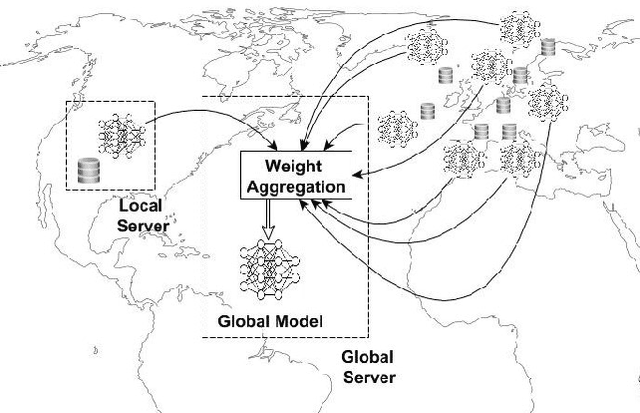
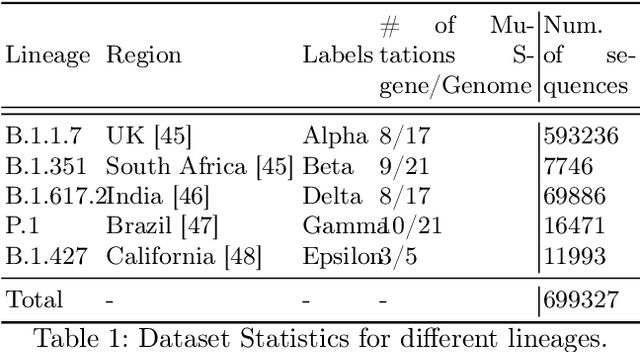
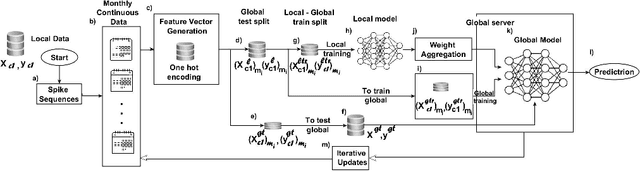
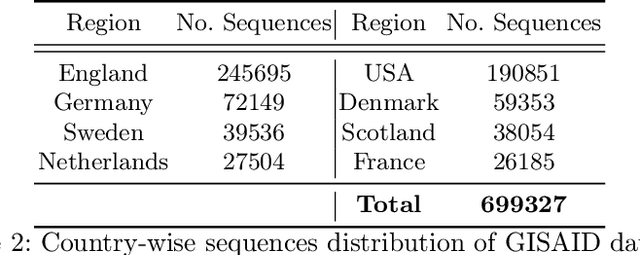
Advancements in genomics technology lead to a rising volume of viral (e.g., SARS-CoV-2) sequence data, resulting in increased usage of machine learning (ML) in bioinformatics. Traditional ML techniques require centralized data collection and processing, posing challenges in realistic healthcare scenarios. Additionally, privacy, ownership, and stringent regulation issues exist when pooling medical data into centralized storage to train a powerful deep learning (DL) model. The Federated learning (FL) approach overcomes such issues by setting up a central aggregator server and a shared global model. It also facilitates data privacy by extracting knowledge while keeping the actual data private. This work proposes a cutting-edge Privacy enhancement through Iterative Collaboration (EPIC) architecture. The network is divided and distributed between local and centralized servers. We demonstrate the EPIC approach to resolve a supervised classification problem to estimate SARS-CoV-2 genomic sequence data lineage without explicitly transferring raw sequence data. We aim to create a universal decentralized optimization framework that allows various data holders to work together and converge to a single predictive model. The findings demonstrate that privacy-preserving strategies can be successfully used with aggregation approaches without materially altering the degree of learning convergence. Finally, we highlight a few potential issues and prospects for study in FL-based approaches to healthcare applications.
MIK: Modified Isolation Kernel for Biological Sequence Visualization, Classification, and Clustering
Oct 21, 2024



The t-Distributed Stochastic Neighbor Embedding (t-SNE) has emerged as a popular dimensionality reduction technique for visualizing high-dimensional data. It computes pairwise similarities between data points by default using an RBF kernel and random initialization (in low-dimensional space), which successfully captures the overall structure but may struggle to preserve the local structure efficiently. This research proposes a novel approach called the Modified Isolation Kernel (MIK) as an alternative to the Gaussian kernel, which is built upon the concept of the Isolation Kernel. MIK uses adaptive density estimation to capture local structures more accurately and integrates robustness measures. It also assigns higher similarity values to nearby points and lower values to distant points. Comparative research using the normal Gaussian kernel, the isolation kernel, and several initialization techniques, including random, PCA, and random walk initializations, are used to assess the proposed approach (MIK). Additionally, we compare the computational efficiency of all $3$ kernels with $3$ different initialization methods. Our experimental results demonstrate several advantages of the proposed kernel (MIK) and initialization method selection. It exhibits improved preservation of the local and global structure and enables better visualization of clusters and subclusters in the embedded space. These findings contribute to advancing dimensionality reduction techniques and provide researchers and practitioners with an effective tool for data exploration, visualization, and analysis in various domains.
Position Specific Scoring Is All You Need? Revisiting Protein Sequence Classification Tasks
Oct 16, 2024


Understanding the structural and functional characteristics of proteins are crucial for developing preventative and curative strategies that impact fields from drug discovery to policy development. An important and popular technique for examining how amino acids make up these characteristics of the protein sequences with position-specific scoring (PSS). While the string kernel is crucial in natural language processing (NLP), it is unclear if string kernels can extract biologically meaningful information from protein sequences, despite the fact that they have been shown to be effective in the general sequence analysis tasks. In this work, we propose a weighted PSS kernel matrix (or W-PSSKM), that combines a PSS representation of protein sequences, which encodes the frequency information of each amino acid in a sequence, with the notion of the string kernel. This results in a novel kernel function that outperforms many other approaches for protein sequence classification. We perform extensive experimentation to evaluate the proposed method. Our findings demonstrate that the W-PSSKM significantly outperforms existing baselines and state-of-the-art methods and achieves up to 45.1\% improvement in classification accuracy.