 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSequence-to-Image Transformation for Sequence Classification Using Rips Complex Construction and Chaos Game Representation
Dec 10, 2025Traditional feature engineering approaches for molecular sequence classification suffer from sparsity issues and computational complexity, while deep learning models often underperform on tabular biological data. This paper introduces a novel topological approach that transforms molecular sequences into images by combining Chaos Game Representation (CGR) with Rips complex construction from algebraic topology. Our method maps sequence elements to 2D coordinates via CGR, computes pairwise distances, and constructs Rips complexes to capture both local structural and global topological features. We provide formal guarantees on representation uniqueness, topological stability, and information preservation. Extensive experiments on anticancer peptide datasets demonstrate superior performance over vector-based, sequence language models, and existing image-based methods, achieving 86.8\% and 94.5\% accuracy on breast and lung cancer datasets, respectively. The topological representation preserves critical sequence information while enabling effective utilization of vision-based deep learning architectures for molecular sequence analysis.
Murmur2Vec: A Hashing Based Solution For Embedding Generation Of COVID-19 Spike Sequences
Dec 10, 2025Early detection and characterization of coronavirus disease (COVID-19), caused by SARS-CoV-2, remain critical for effective clinical response and public-health planning. The global availability of large-scale viral sequence data presents significant opportunities for computational analysis; however, existing approaches face notable limitations. Phylogenetic tree-based methods are computationally intensive and do not scale efficiently to today's multi-million-sequence datasets. Similarly, current embedding-based techniques often rely on aligned sequences or exhibit suboptimal predictive performance and high runtime costs, creating barriers to practical large-scale analysis. In this study, we focus on the most prevalent SARS-CoV-2 lineages associated with the spike protein region and introduce a scalable embedding method that leverages hashing to generate compact, low-dimensional representations of spike sequences. These embeddings are subsequently used to train a variety of machine learning models for supervised lineage classification. We conduct an extensive evaluation comparing our approach with multiple baseline and state-of-the-art biological sequence embedding methods across diverse metrics. Our results demonstrate that the proposed embeddings offer substantial improvements in efficiency, achieving up to 86.4\% classification accuracy while reducing embedding generation time by as much as 99.81\%. This highlights the method's potential as a fast, effective, and scalable solution for large-scale viral sequence analysis.
Sequence Analysis Using the Bezier Curve
Mar 18, 2025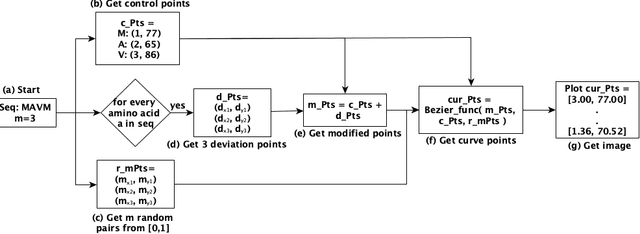
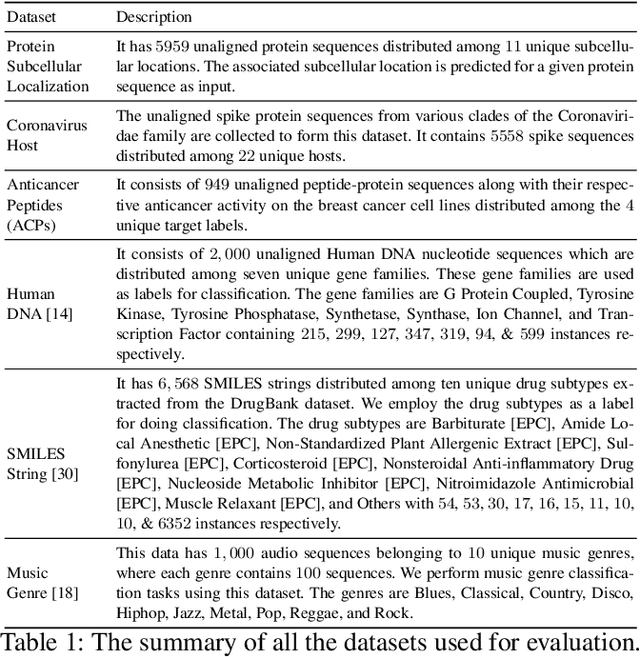

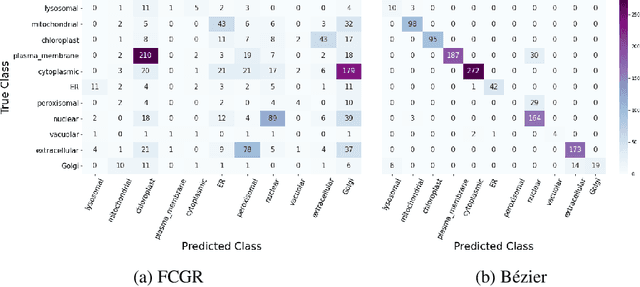
The analysis of sequences (e.g., protein, DNA, and SMILES string) is essential for disease diagnosis, biomaterial engineering, genetic engineering, and drug discovery domains. Conventional analytical methods focus on transforming sequences into numerical representations for applying machine learning/deep learning-based sequence characterization. However, their efficacy is constrained by the intrinsic nature of deep learning (DL) models, which tend to exhibit suboptimal performance when applied to tabular data. An alternative group of methodologies endeavors to convert biological sequences into image forms by applying the concept of Chaos Game Representation (CGR). However, a noteworthy drawback of these methods lies in their tendency to map individual elements of the sequence onto a relatively small subset of designated pixels within the generated image. The resulting sparse image representation may not adequately encapsulate the comprehensive sequence information, potentially resulting in suboptimal predictions. In this study, we introduce a novel approach to transform sequences into images using the B\'ezier curve concept for element mapping. Mapping the elements onto a curve enhances the sequence information representation in the respective images, hence yielding better DL-based classification performance. We employed different sequence datasets to validate our system by using different classification tasks, and the results illustrate that our B\'ezier curve method is able to achieve good performance for all the tasks.
Position Specific Scoring Is All You Need? Revisiting Protein Sequence Classification Tasks
Oct 16, 2024


Understanding the structural and functional characteristics of proteins are crucial for developing preventative and curative strategies that impact fields from drug discovery to policy development. An important and popular technique for examining how amino acids make up these characteristics of the protein sequences with position-specific scoring (PSS). While the string kernel is crucial in natural language processing (NLP), it is unclear if string kernels can extract biologically meaningful information from protein sequences, despite the fact that they have been shown to be effective in the general sequence analysis tasks. In this work, we propose a weighted PSS kernel matrix (or W-PSSKM), that combines a PSS representation of protein sequences, which encodes the frequency information of each amino acid in a sequence, with the notion of the string kernel. This results in a novel kernel function that outperforms many other approaches for protein sequence classification. We perform extensive experimentation to evaluate the proposed method. Our findings demonstrate that the W-PSSKM significantly outperforms existing baselines and state-of-the-art methods and achieves up to 45.1\% improvement in classification accuracy.
DANCE: Deep Learning-Assisted Analysis of Protein Sequences Using Chaos Enhanced Kaleidoscopic Images
Sep 10, 2024Cancer is a complex disease characterized by uncontrolled cell growth. T cell receptors (TCRs), crucial proteins in the immune system, play a key role in recognizing antigens, including those associated with cancer. Recent advancements in sequencing technologies have facilitated comprehensive profiling of TCR repertoires, uncovering TCRs with potent anti-cancer activity and enabling TCR-based immunotherapies. However, analyzing these intricate biomolecules necessitates efficient representations that capture their structural and functional information. T-cell protein sequences pose unique challenges due to their relatively smaller lengths compared to other biomolecules. An image-based representation approach becomes a preferred choice for efficient embeddings, allowing for the preservation of essential details and enabling comprehensive analysis of T-cell protein sequences. In this paper, we propose to generate images from the protein sequences using the idea of Chaos Game Representation (CGR) using the Kaleidoscopic images approach. This Deep Learning Assisted Analysis of Protein Sequences Using Chaos Enhanced Kaleidoscopic Images (called DANCE) provides a unique way to visualize protein sequences by recursively applying chaos game rules around a central seed point. we perform the classification of the T cell receptors (TCRs) protein sequences in terms of their respective target cancer cells, as TCRs are known for their immune response against cancer disease. The TCR sequences are converted into images using the DANCE method. We employ deep-learning vision models to perform the classification to obtain insights into the relationship between the visual patterns observed in the generated kaleidoscopic images and the underlying protein properties. By combining CGR-based image generation with deep learning classification, this study opens novel possibilities in the protein analysis domain.
T Cell Receptor Protein Sequences and Sparse Coding: A Novel Approach to Cancer Classification
Apr 25, 2023
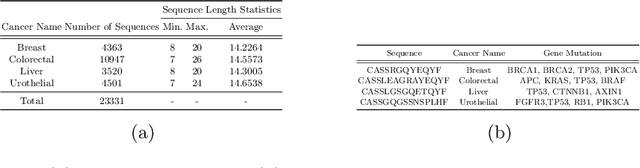
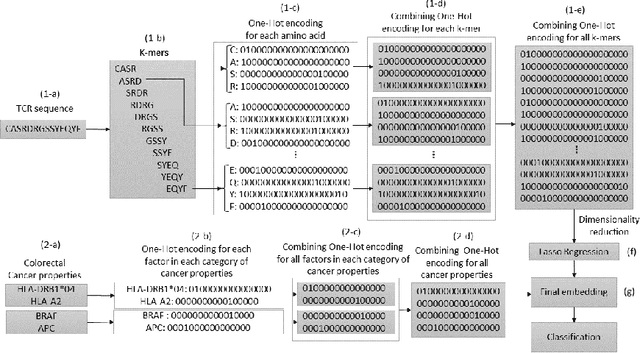
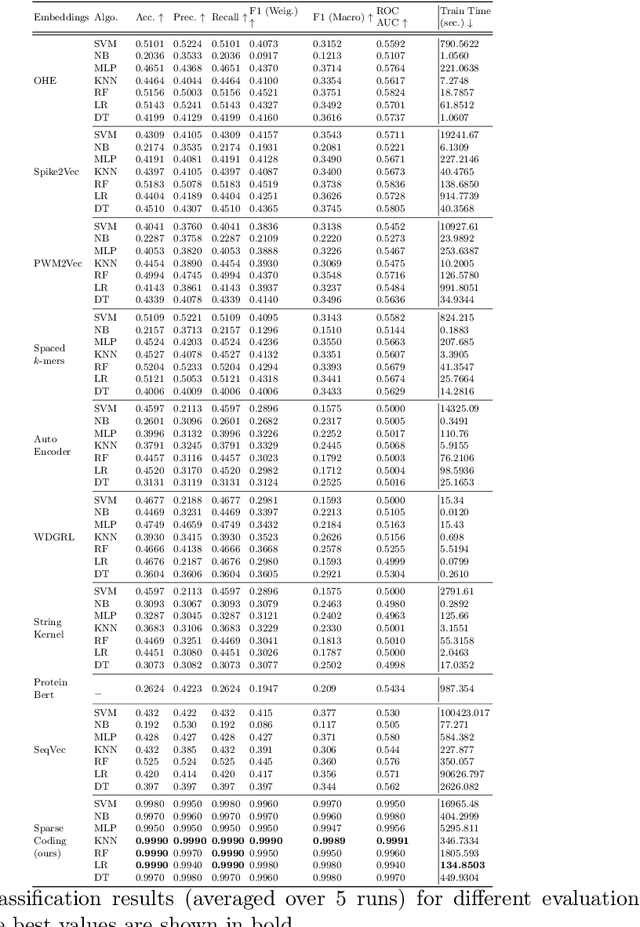
Cancer is a complex disease characterized by uncontrolled cell growth and proliferation. T cell receptors (TCRs) are essential proteins for the adaptive immune system, and their specific recognition of antigens plays a crucial role in the immune response against diseases, including cancer. The diversity and specificity of TCRs make them ideal for targeting cancer cells, and recent advancements in sequencing technologies have enabled the comprehensive profiling of TCR repertoires. This has led to the discovery of TCRs with potent anti-cancer activity and the development of TCR-based immunotherapies. In this study, we investigate the use of sparse coding for the multi-class classification of TCR protein sequences with cancer categories as target labels. Sparse coding is a popular technique in machine learning that enables the representation of data with a set of informative features and can capture complex relationships between amino acids and identify subtle patterns in the sequence that might be missed by low-dimensional methods. We first compute the k-mers from the TCR sequences and then apply sparse coding to capture the essential features of the data. To improve the predictive performance of the final embeddings, we integrate domain knowledge regarding different types of cancer properties. We then train different machine learning (linear and non-linear) classifiers on the embeddings of TCR sequences for the purpose of supervised analysis. Our proposed embedding method on a benchmark dataset of TCR sequences significantly outperforms the baselines in terms of predictive performance, achieving an accuracy of 99.8\%. Our study highlights the potential of sparse coding for the analysis of TCR protein sequences in cancer research and other related fields.
PCD2Vec: A Poisson Correction Distance-Based Approach for Viral Host Classification
Apr 13, 2023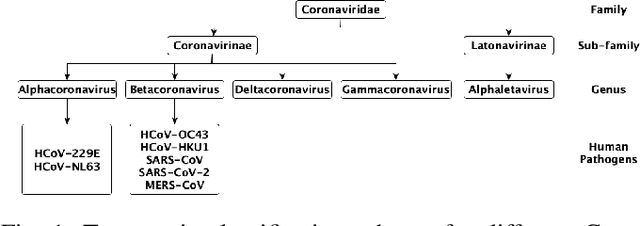
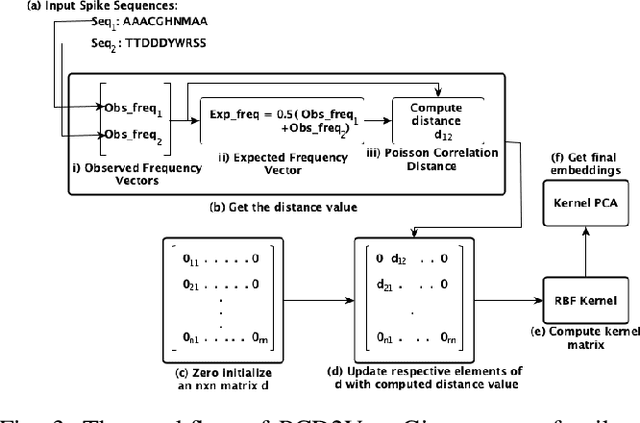
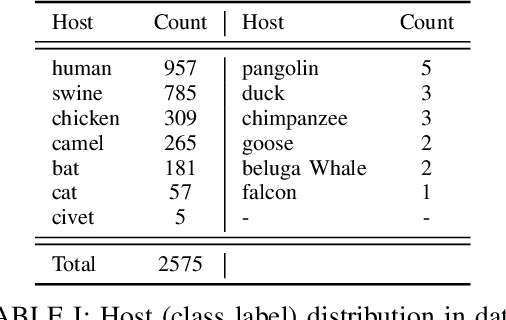
Coronaviruses are membrane-enveloped, non-segmented positive-strand RNA viruses belonging to the Coronaviridae family. Various animal species, mainly mammalian and avian, are severely infected by various coronaviruses, causing serious concerns like the recent pandemic (COVID-19). Therefore, building a deeper understanding of these viruses is essential to devise prevention and mitigation mechanisms. In the Coronavirus genome, an essential structural region is the spike region, and it's responsible for attaching the virus to the host cell membrane. Therefore, the usage of only the spike protein, instead of the full genome, provides most of the essential information for performing analyses such as host classification. In this paper, we propose a novel method for predicting the host specificity of coronaviruses by analyzing spike protein sequences from different viral subgenera and species. Our method involves using the Poisson correction distance to generate a distance matrix, followed by using a radial basis function (RBF) kernel and kernel principal component analysis (PCA) to generate a low-dimensional embedding. Finally, we apply classification algorithms to the low-dimensional embedding to generate the resulting predictions of the host specificity of coronaviruses. We provide theoretical proofs for the non-negativity, symmetry, and triangle inequality properties of the Poisson correction distance metric, which are important properties in a machine-learning setting. By encoding the spike protein structure and sequences using this comprehensive approach, we aim to uncover hidden patterns in the biological sequences to make accurate predictions about host specificity. Finally, our classification results illustrate that our method can achieve higher predictive accuracy and improve performance over existing baselines.
Exploring The Potential Of GANs In Biological Sequence Analysis
Mar 04, 2023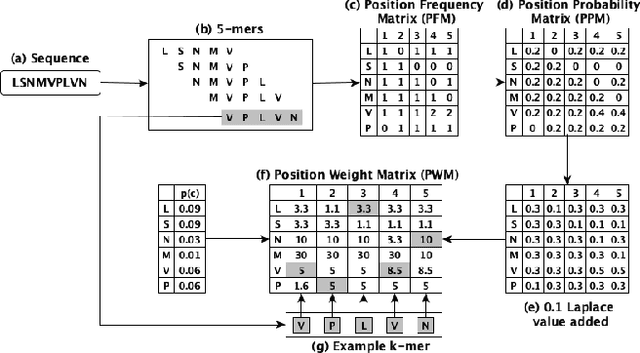
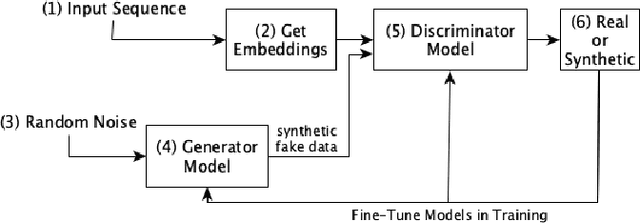
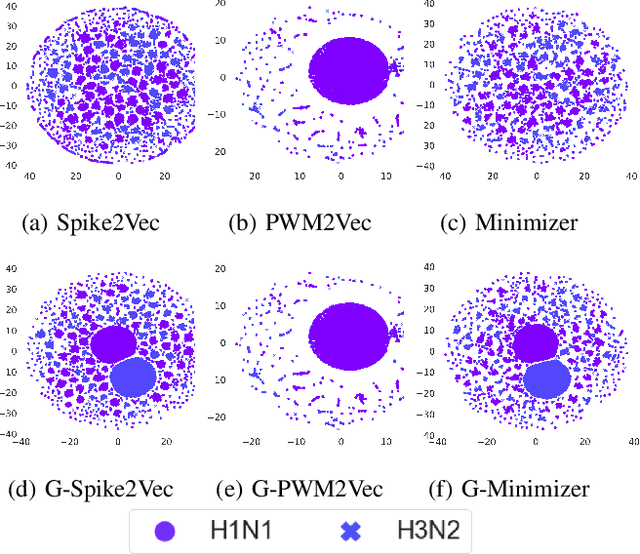
Biological sequence analysis is an essential step toward building a deeper understanding of the underlying functions, structures, and behaviors of the sequences. It can help in identifying the characteristics of the associated organisms, like viruses, etc., and building prevention mechanisms to eradicate their spread and impact, as viruses are known to cause epidemics that can become pandemics globally. New tools for biological sequence analysis are provided by machine learning (ML) technologies to effectively analyze the functions and structures of the sequences. However, these ML-based methods undergo challenges with data imbalance, generally associated with biological sequence datasets, which hinders their performance. Although various strategies are present to address this issue, like the SMOTE algorithm, which creates synthetic data, however, they focus on local information rather than the overall class distribution. In this work, we explore a novel approach to handle the data imbalance issue based on Generative Adversarial Networks (GANs) which use the overall data distribution. GANs are utilized to generate synthetic data that closely resembles the real one, thus this generated data can be employed to enhance the ML models' performance by eradicating the class imbalance problem for biological sequence analysis. We perform 3 distinct classification tasks by using 3 different sequence datasets (Influenza A Virus, PALMdb, VDjDB) and our results illustrate that GANs can improve the overall classification performance.
Efficient Classification of SARS-CoV-2 Spike Sequences Using Federated Learning
Feb 17, 2023



This paper presents a federated learning (FL) approach to train an AI model for SARS-Cov-2 coronavirus variant identification. We analyze the SARS-CoV-2 spike sequences in a distributed way, without data sharing, to detect different variants of the rapidly mutating coronavirus. A vast amount of sequencing data of SARS-CoV-2 is available due to various genomic monitoring initiatives by several nations. However, privacy concerns involving patient health information and national public health conditions could hinder openly sharing this data. In this work, we propose a lightweight FL paradigm to cooperatively analyze the spike protein sequences of SARS-CoV-2 privately, using the locally stored data to train a prediction model from remote nodes. Our method maintains the confidentiality of local data (that could be stored in different locations) yet allows us to reliably detect and identify different known and unknown variants of the novel coronavirus SARS-CoV-2. We compare the performance of our approach on spike sequence data with the recently proposed state-of-the-art methods for classification from spike sequences. Using the proposed approach, we achieve an overall accuracy of $93\%$ on the coronavirus variant identification task. To the best of our knowledge, this is the first work in the federated learning paradigm for biological sequence analysis. Since the proposed model is distributed in nature, it could scale on ``Big Data'' easily. We plan to use this proof-of-concept to implement a privacy-preserving pandemic response strategy.