 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Hierarchical Feature Engineering Framework for Automated Classification of Phonotraumatic and Non-Phonotraumatic Vocal Hyperfunction
Jun 04, 2026Ambulatory neck-surface acceleration enables non-invasive monitoring of vocal hyperfunction, yet robust biomarkers for its subtypes remain limited. This study investigates the NeckVibe Challenge dataset to distinguish phonotraumatic (PVH) and non-phonotraumatic (NPVH) from healthy controls. We propose a hierarchical feature engineering framework comprising: (i) static, (ii) dynamic, (iii) ratio-based, (iv) coupling features capturing source filter interactions. While univariate statistical analysis shows strong separability for PVH but limited significance for NPVH, our machine learning pipeline, tailored for high-dimensional feature integration, identifies that coupling features are crucial for both tasks. We achieve an AUC of 0.891 for PVH and 0.728 for NPVH, suggesting that while PVH is near-linearly separable, NPVH discrimination benefits from modeling non-linear feature interactions.
GrowTAS: Progressive Expansion from Small to Large Subnets for Efficient ViT Architecture Search
Dec 13, 2025Transformer architecture search (TAS) aims to automatically discover efficient vision transformers (ViTs), reducing the need for manual design. Existing TAS methods typically train an over-parameterized network (i.e., a supernet) that encompasses all candidate architectures (i.e., subnets). However, all subnets share the same set of weights, which leads to interference that degrades the smaller subnets severely. We have found that well-trained small subnets can serve as a good foundation for training larger ones. Motivated by this, we propose a progressive training framework, dubbed GrowTAS, that begins with training small subnets and incorporate larger ones gradually. This enables reducing the interference and stabilizing a training process. We also introduce GrowTAS+ that fine-tunes a subset of weights only to further enhance the performance of large subnets. Extensive experiments on ImageNet and several transfer learning benchmarks, including CIFAR-10/100, Flowers, CARS, and INAT-19, demonstrate the effectiveness of our approach over current TAS methods
MoCoRP: Modeling Consistent Relations between Persona and Response for Persona-based Dialogue
Dec 08, 2025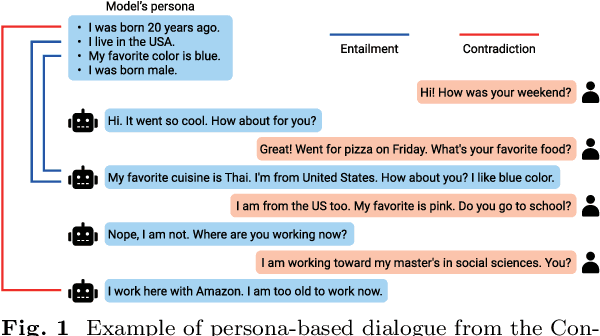



As dialogue systems become increasingly important across various domains, a key challenge in persona-based dialogue is generating engaging and context-specific interactions while ensuring the model acts with a coherent personality. However, existing persona-based dialogue datasets lack explicit relations between persona sentences and responses, which makes it difficult for models to effectively capture persona information. To address these issues, we propose MoCoRP (Modeling Consistent Relations between Persona and Response), a framework that incorporates explicit relations into language models. MoCoRP leverages an NLI expert to explicitly extract the NLI relations between persona sentences and responses, enabling the model to effectively incorporate appropriate persona information from the context into its responses. We applied this framework to pre-trained models like BART and further extended it to modern large language models (LLMs) through alignment tuning. Experimental results on the public datasets ConvAI2 and MPChat demonstrate that MoCoRP outperforms existing baselines, achieving superior persona consistency and engaging, context-aware dialogue generation. Furthermore, our model not only excels in quantitative metrics but also shows significant improvements in qualitative aspects. These results highlight the effectiveness of explicitly modeling persona-response relations in persona-based dialogue. The source codes of MoCoRP are available at https://github.com/DMCB-GIST/MoCoRP.
Quantum Supremacy in Tomographic Imaging: Advances in Quantum Tomography Algorithms
Feb 07, 2025Quantum computing has emerged as a transformative paradigm, capable of tackling complex computational problems that are infeasible for classical methods within a practical timeframe. At the core of this advancement lies the concept of quantum supremacy, which signifies the ability of quantum processors to surpass classical systems in specific tasks. In the context of tomographic image reconstruction, quantum optimization algorithms enable faster processing and clearer imaging than conventional methods. This study further substantiates quantum supremacy by reducing the required projection angles for tomographic reconstruction while enhancing robustness against image artifacts. Notably, our experiments demonstrated that the proposed algorithm accurately reconstructed tomographic images without artifacts, even when up to 50% error was introduced into the sinogram to induce ring artifacts. Furthermore, it achieved precise reconstructions using only 50% of the projection angles from the original sinogram spanning 0{\deg} to 180{\deg}. These findings highlight the potential of quantum algorithms to revolutionize tomographic imaging by enabling efficient and accurate reconstructions under challenging conditions, paving the way for broader applications in medical imaging, material science, and advanced tomography systems as quantum computing technologies continue to advance.
Efficient Few-Shot Neural Architecture Search by Counting the Number of Nonlinear Functions
Dec 19, 2024Neural architecture search (NAS) enables finding the best-performing architecture from a search space automatically. Most NAS methods exploit an over-parameterized network (i.e., a supernet) containing all possible architectures (i.e., subnets) in the search space. However, the subnets that share the same set of parameters are likely to have different characteristics, interfering with each other during training. To address this, few-shot NAS methods have been proposed that divide the space into a few subspaces and employ a separate supernet for each subspace to limit the extent of weight sharing. They achieve state-of-the-art performance, but the computational cost increases accordingly. We introduce in this paper a novel few-shot NAS method that exploits the number of nonlinear functions to split the search space. To be specific, our method divides the space such that each subspace consists of subnets with the same number of nonlinear functions. Our splitting criterion is efficient, since it does not require comparing gradients of a supernet to split the space. In addition, we have found that dividing the space allows us to reduce the channel dimensions required for each supernet, which enables training multiple supernets in an efficient manner. We also introduce a supernet-balanced sampling (SBS) technique, sampling several subnets at each training step, to train different supernets evenly within a limited number of training steps. Extensive experiments on standard NAS benchmarks demonstrate the effectiveness of our approach. Our code is available at https://cvlab.yonsei.ac.kr/projects/EFS-NAS.
Illustrious: an Open Advanced Illustration Model
Sep 30, 2024



In this work, we share the insights for achieving state-of-the-art quality in our text-to-image anime image generative model, called Illustrious. To achieve high resolution, dynamic color range images, and high restoration ability, we focus on three critical approaches for model improvement. First, we delve into the significance of the batch size and dropout control, which enables faster learning of controllable token based concept activations. Second, we increase the training resolution of images, affecting the accurate depiction of character anatomy in much higher resolution, extending its generation capability over 20MP with proper methods. Finally, we propose the refined multi-level captions, covering all tags and various natural language captions as a critical factor for model development. Through extensive analysis and experiments, Illustrious demonstrates state-of-the-art performance in terms of animation style, outperforming widely-used models in illustration domains, propelling easier customization and personalization with nature of open source. We plan to publicly release updated Illustrious model series sequentially as well as sustainable plans for improvements.
Deep Metric Loss for Multimodal Learning
Aug 21, 2023Multimodal learning often outperforms its unimodal counterparts by exploiting unimodal contributions and cross-modal interactions. However, focusing only on integrating multimodal features into a unified comprehensive representation overlooks the unimodal characteristics. In real data, the contributions of modalities can vary from instance to instance, and they often reinforce or conflict with each other. In this study, we introduce a novel \text{MultiModal} loss paradigm for multimodal learning, which subgroups instances according to their unimodal contributions. \text{MultiModal} loss can prevent inefficient learning caused by overfitting and efficiently optimize multimodal models. On synthetic data, \text{MultiModal} loss demonstrates improved classification performance by subgrouping difficult instances within certain modalities. On four real multimodal datasets, our loss is empirically shown to improve the performance of recent models. Ablation studies verify the effectiveness of our loss. Additionally, we show that our loss generates a reliable prediction score for each modality, which is essential for subgrouping. Our \text{MultiModal} loss is a novel loss function to subgroup instances according to the contribution of modalities in multimodal learning and is applicable to a variety of multimodal models with unimodal decisions. Our code is available at https://github.com/SehwanMoon/MultiModalLoss.
PINNet: a deep neural network with pathway prior knowledge for Alzheimer's disease
Nov 27, 2022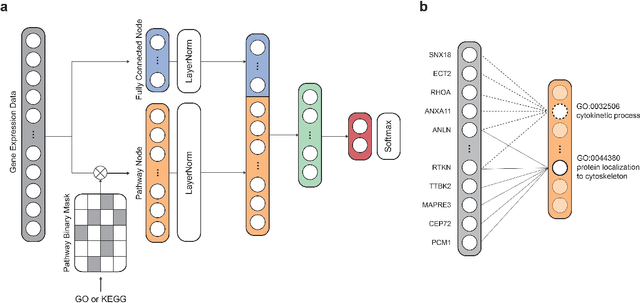

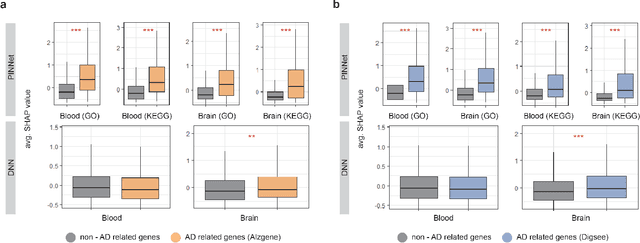
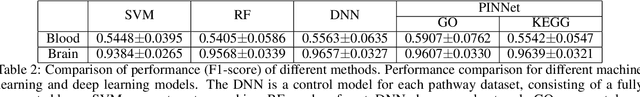
Identification of Alzheimer's Disease (AD)-related transcriptomic signatures from blood is important for early diagnosis of the disease. Deep learning techniques are potent classifiers for AD diagnosis, but most have been unable to identify biomarkers because of their lack of interpretability. To address these challenges, we propose a pathway information-based neural network (PINNet) to predict AD patients and analyze blood and brain transcriptomic signatures using an interpretable deep learning model. PINNet is a deep neural network (DNN) model with pathway prior knowledge from either the Gene Ontology or Kyoto Encyclopedia of Genes and Genomes databases. Then, a backpropagation-based model interpretation method was applied to reveal essential pathways and genes for predicting AD. We compared the performance of PINNet with a DNN model without a pathway. Performances of PINNet outperformed or were similar to those of DNN without a pathway using blood and brain gene expressions, respectively. Moreover, PINNet considers more AD-related genes as essential features than DNN without a pathway in the learning process. Pathway analysis of protein-protein interaction modules of highly contributed genes showed that AD-related genes in blood were enriched with cell migration, PI3K-Akt, MAPK signaling, and apoptosis in blood. The pathways enriched in the brain module included cell migration, PI3K-Akt, MAPK signaling, apoptosis, protein ubiquitination, and t-cell activation. Collectively, with prior knowledge about pathways, PINNet reveals essential pathways related to AD.
SDGCCA: Supervised Deep Generalized Canonical Correlation Analysis for Multi-omics Integration
Apr 17, 2022
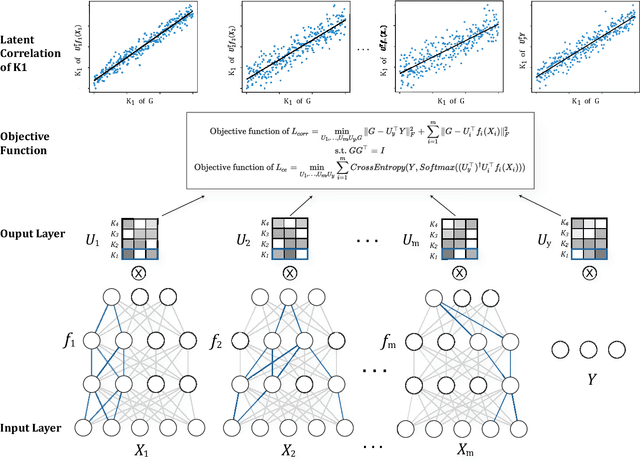
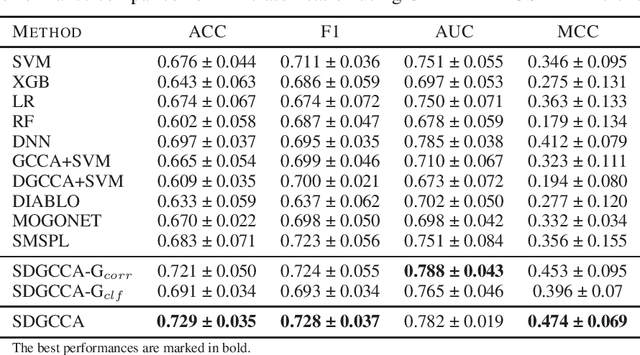
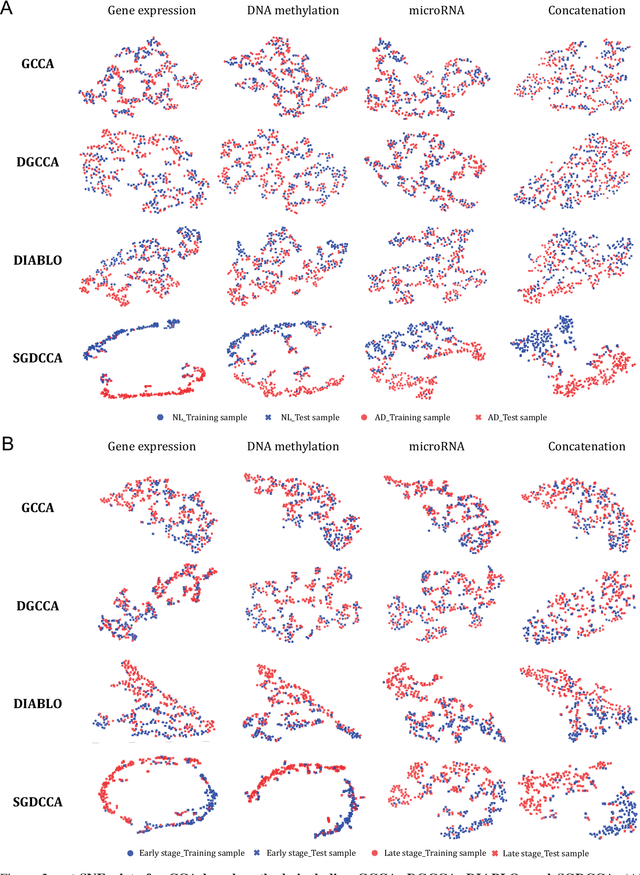
Integration of multi-omics data provides opportunities for revealing biological mechanisms related to certain phenotypes. We propose a novel method of multi-omics integration called supervised deep generalized canonical correlation analysis (SDGCCA) for modeling correlation structures between nonlinear multi-omics manifolds, aiming for improving classification of phenotypes and revealing biomarkers related to phenotypes. SDGCCA addresses the limitations of other canonical correlation analysis (CCA)-based models (e.g., deep CCA, deep generalized CCA) by considering complex/nonlinear cross-data correlations and discriminating phenotype groups. Although there are a few methods for nonlinear CCA projections for discriminant purposes of phenotypes, they only consider two views. On the other hand, SDGCCA is the nonlinear multiview CCA projection method for discrimination. When we applied SDGCCA to prediction of patients of Alzheimer's disease (AD) and discrimination of early- and late-stage cancers, it outperformed other CCA-based methods and other supervised methods. In addition, we demonstrate that SDGCCA can be used for feature selection to identify important multi-omics biomarkers. In the application on AD data, SDGCCA identified clusters of genes in multi-omics data, which are well known to be associated with AD.
A molecular generative model with genetic algorithm and tree search for cancer samples
Dec 16, 2021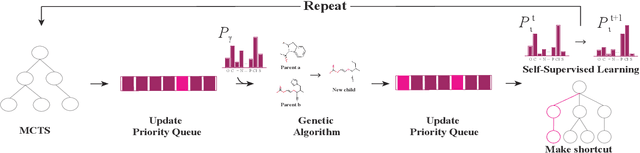
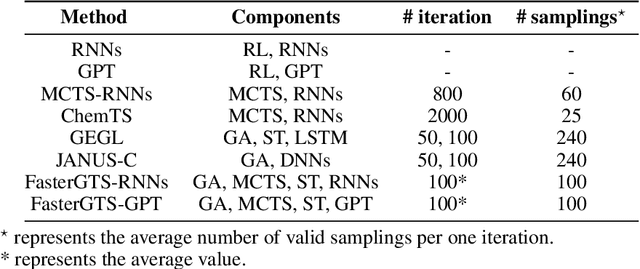
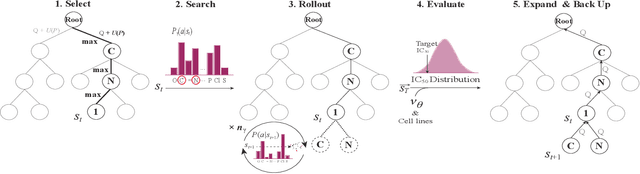
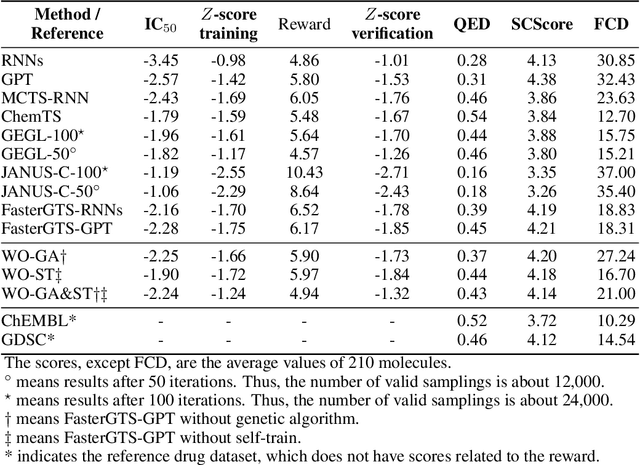
Personalized medicine is expected to maximize the intended drug effects and minimize side effects by treating patients based on their genetic profiles. Thus, it is important to generate drugs based on the genetic profiles of diseases, especially in anticancer drug discovery. However, this is challenging because the vast chemical space and variations in cancer properties require a huge time resource to search for proper molecules. Therefore, an efficient and fast search method considering genetic profiles is required for de novo molecular design of anticancer drugs. Here, we propose a faster molecular generative model with genetic algorithm and tree search for cancer samples (FasterGTS). FasterGTS is constructed with a genetic algorithm and a Monte Carlo tree search with three deep neural networks: supervised learning, self-trained, and value networks, and it generates anticancer molecules based on the genetic profiles of a cancer sample. When compared to other methods, FasterGTS generated cancer sample-specific molecules with general chemical properties required for cancer drugs within the limited numbers of samplings. We expect that FasterGTS contributes to the anticancer drug generation.