 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBMFM-RNA: An Open Framework for Building and Evaluating Transcriptomic Foundation Models
Jun 17, 2025



Transcriptomic foundation models (TFMs) have recently emerged as powerful tools for analyzing gene expression in cells and tissues, supporting key tasks such as cell-type annotation, batch correction, and perturbation prediction. However, the diversity of model implementations and training strategies across recent TFMs, though promising, makes it challenging to isolate the contribution of individual design choices or evaluate their potential synergies. This hinders the field's ability to converge on best practices and limits the reproducibility of insights across studies. We present BMFM-RNA, an open-source, modular software package that unifies diverse TFM pretraining and fine-tuning objectives within a single framework. Leveraging this capability, we introduce a novel training objective, whole cell expression decoder (WCED), which captures global expression patterns using an autoencoder-like CLS bottleneck representation. In this paper, we describe the framework, supported input representations, and training objectives. We evaluated four model checkpoints pretrained on CELLxGENE using combinations of masked language modeling (MLM), WCED and multitask learning. Using the benchmarking capabilities of BMFM-RNA, we show that WCED-based models achieve performance that matches or exceeds state-of-the-art approaches like scGPT across more than a dozen datasets in both zero-shot and fine-tuning tasks. BMFM-RNA, available as part of the biomed-multi-omics project ( https://github.com/BiomedSciAI/biomed-multi-omic ), offers a reproducible foundation for systematic benchmarking and community-driven exploration of optimal TFM training strategies, enabling the development of more effective tools to leverage the latest advances in AI for understanding cell biology.
Does your model understand genes? A benchmark of gene properties for biological and text models
Dec 05, 2024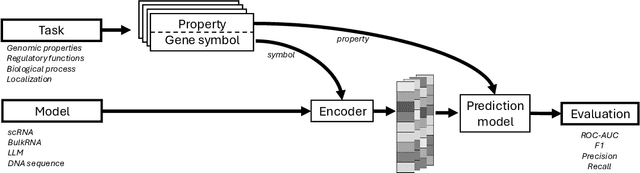
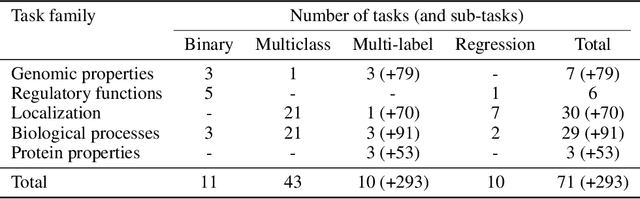
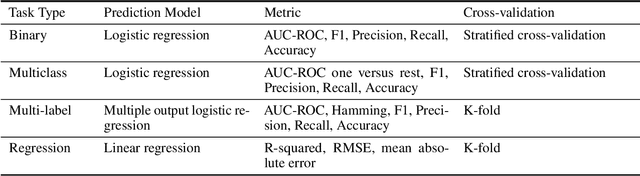

The application of deep learning methods, particularly foundation models, in biological research has surged in recent years. These models can be text-based or trained on underlying biological data, especially omics data of various types. However, comparing the performance of these models consistently has proven to be a challenge due to differences in training data and downstream tasks. To tackle this problem, we developed an architecture-agnostic benchmarking approach that, instead of evaluating the models directly, leverages entity representation vectors from each model and trains simple predictive models for each benchmarking task. This ensures that all types of models are evaluated using the same input and output types. Here we focus on gene properties collected from professionally curated bioinformatics databases. These gene properties are categorized into five major groups: genomic properties, regulatory functions, localization, biological processes, and protein properties. Overall, we define hundreds of tasks based on these databases, which include binary, multi-label, and multi-class classification tasks. We apply these benchmark tasks to evaluate expression-based models, large language models, protein language models, DNA-based models, and traditional baselines. Our findings suggest that text-based models and protein language models generally outperform expression-based models in genomic properties and regulatory functions tasks, whereas expression-based models demonstrate superior performance in localization tasks. These results should aid in the development of more informed artificial intelligence strategies for biological understanding and therapeutic discovery. To ensure the reproducibility and transparency of our findings, we have made the source code and benchmark data publicly accessible for further investigation and expansion at github.com/BiomedSciAI/gene-benchmark.
Hierarchical Bias-Driven Stratification for Interpretable Causal Effect Estimation
Jan 31, 2024Interpretability and transparency are essential for incorporating causal effect models from observational data into policy decision-making. They can provide trust for the model in the absence of ground truth labels to evaluate the accuracy of such models. To date, attempts at transparent causal effect estimation consist of applying post hoc explanation methods to black-box models, which are not interpretable. Here, we present BICauseTree: an interpretable balancing method that identifies clusters where natural experiments occur locally. Our approach builds on decision trees with a customized objective function to improve balancing and reduce treatment allocation bias. Consequently, it can additionally detect subgroups presenting positivity violations, exclude them, and provide a covariate-based definition of the target population we can infer from and generalize to. We evaluate the method's performance using synthetic and realistic datasets, explore its bias-interpretability tradeoff, and show that it is comparable with existing approaches.
Propensity score models are better when post-calibrated
Nov 02, 2022Theoretical guarantees for causal inference using propensity scores are partly based on the scores behaving like conditional probabilities. However, scores between zero and one, especially when outputted by flexible statistical estimators, do not necessarily behave like probabilities. We perform a simulation study to assess the error in estimating the average treatment effect before and after applying a simple and well-established post-processing method to calibrate the propensity scores. We find that post-calibration reduces the error in effect estimation for expressive uncalibrated statistical estimators, and that this improvement is not mediated by better balancing. The larger the initial lack of calibration, the larger the improvement in effect estimation, with the effect on already-calibrated estimators being very small. Given the improvement in effect estimation and that post-calibration is computationally cheap, we recommend it will be adopted when modelling propensity scores with expressive models.
A discriminative approach for finding and characterizing positivity violations using decision trees
Jul 18, 2019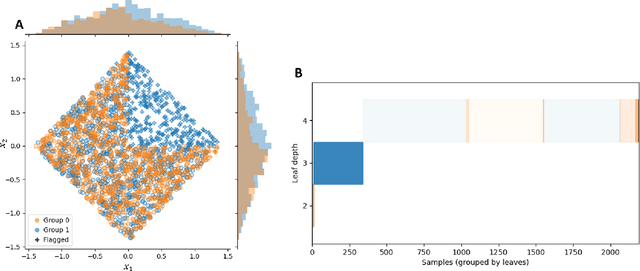
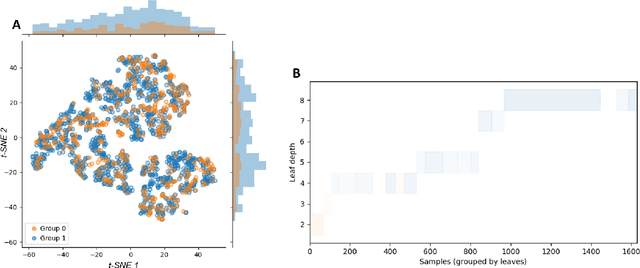
The assumption of positivity in causal inference (also known as common support and co-variate overlap) is necessary to obtain valid causal estimates. Therefore, confirming it holds in a given dataset is an important first step of any causal analysis. Most common methods to date are insufficient for discovering non-positivity, as they do not scale for modern high-dimensional covariate spaces, or they cannot pinpoint the subpopulation violating positivity. To overcome these issues, we suggest to harness decision trees for detecting violations. By dividing the covariate space into mutually exclusive regions, each with maximized homogeneity of treatment groups, decision trees can be used to automatically detect subspaces violating positivity. By augmenting the method with an additional random forest model, we can quantify the robustness of the violation within each subspace. This solution is scalable and provides an interpretable characterization of the subspaces in which violations occur. We provide a visualization of the stratification rules that define each subpopulation, combined with the severity of positivity violation within it. We also provide an interactive version of the visualization that allows a deeper dive into the properties of each subspace.
An Evaluation Toolkit to Guide Model Selection and Cohort Definition in Causal Inference
Jun 02, 2019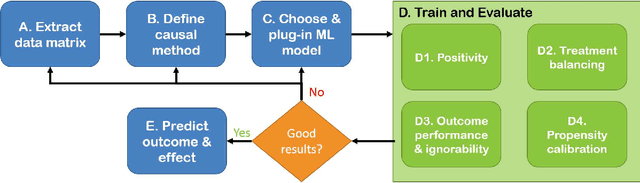

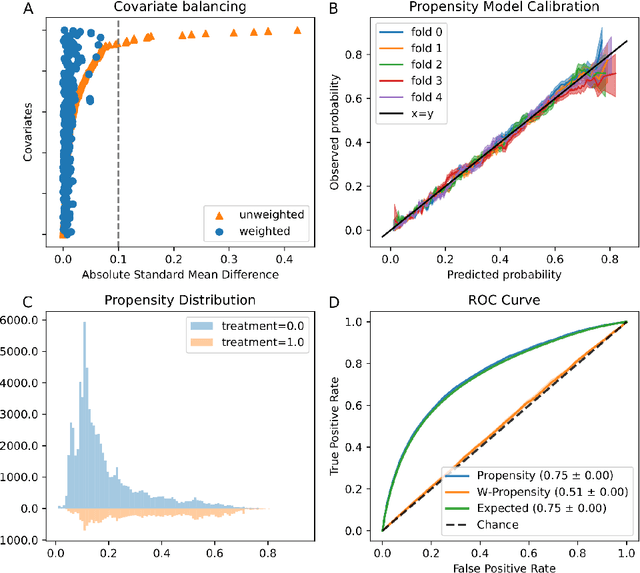
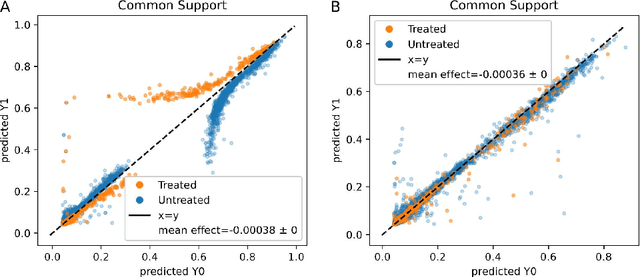
Real world observational data, together with causal inference, allow the estimation of causal effects when randomized controlled trials are not available. To be accepted into practice, such predictive models must be validated for the dataset at hand, and thus require a comprehensive evaluation toolkit, as introduced here. Since effect estimation cannot be evaluated directly, we turn to evaluating the various observable properties of causal inference, namely the observed outcome and treatment assignment. We developed a toolkit that expands established machine learning evaluation methods and adds several causal-specific ones. Evaluations can be applied in cross-validation, in a train-test scheme, or on the training data. Multiple causal inference methods are implemented within the toolkit in a way that allows modular use of the underlying machine learning models. Thus, the toolkit is agnostic to the machine learning model that is used. We showcase our approach using a rheumatoid arthritis cohort (consisting of about 120K patients) extracted from the IBM MarketScan(R) Research Database. We introduce an iterative pipeline of data definition, model definition, and model evaluation. Using this pipeline, we demonstrate how each of the evaluation components helps drive model selection and refinement of data extraction criteria in a way that provides more reproducible results and ensures that the causal question is answerable with available data. Furthermore, we show how the evaluation toolkit can be used to ensure that performance is maintained when applied to subsets of the data, thus allowing exploration of questions that move towards personalized medicine.
Benchmarking Framework for Performance-Evaluation of Causal Inference Analysis
Mar 20, 2018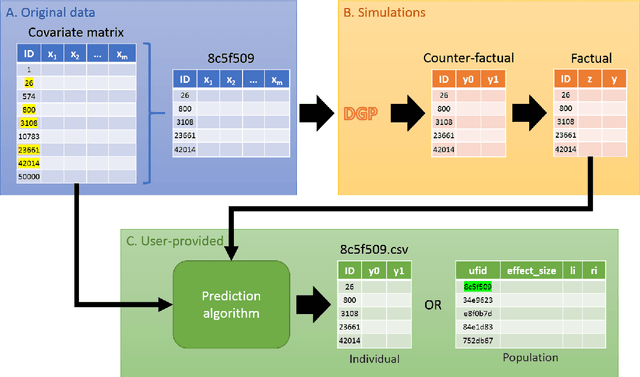
Causal inference analysis is the estimation of the effects of actions on outcomes. In the context of healthcare data this means estimating the outcome of counter-factual treatments (i.e. including treatments that were not observed) on a patient's outcome. Compared to classic machine learning methods, evaluation and validation of causal inference analysis is more challenging because ground truth data of counter-factual outcome can never be obtained in any real-world scenario. Here, we present a comprehensive framework for benchmarking algorithms that estimate causal effect. The framework includes unlabeled data for prediction, labeled data for validation, and code for automatic evaluation of algorithm predictions using both established and novel metrics. The data is based on real-world covariates, and the treatment assignments and outcomes are based on simulations, which provides the basis for validation. In this framework we address two questions: one of scaling, and the other of data-censoring. The framework is available as open source code at https://github.com/IBM-HRL-MLHLS/IBM-Causal-Inference-Benchmarking-Framework