 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIncorporating intratumoral heterogeneity into weakly-supervised deep learning models via variance pooling
Jun 17, 2022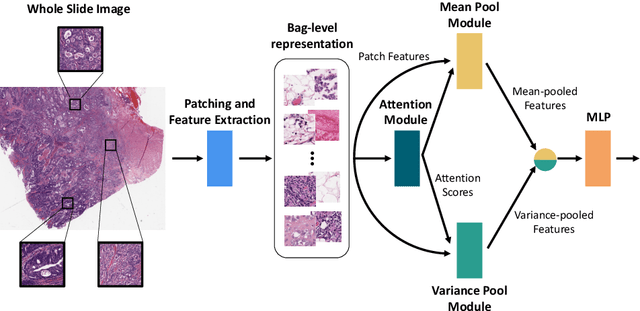
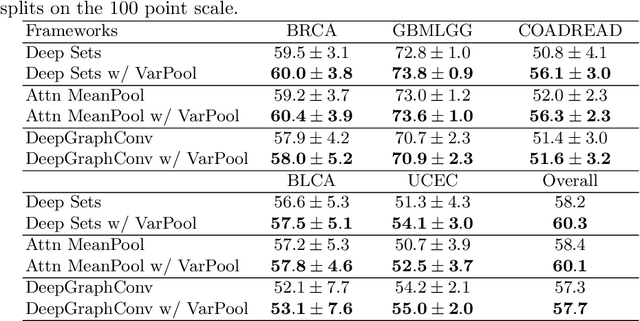
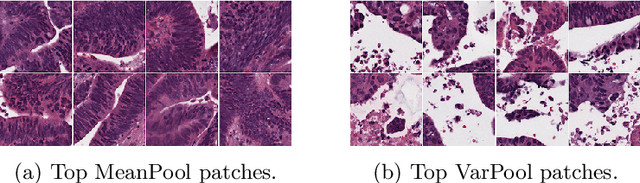
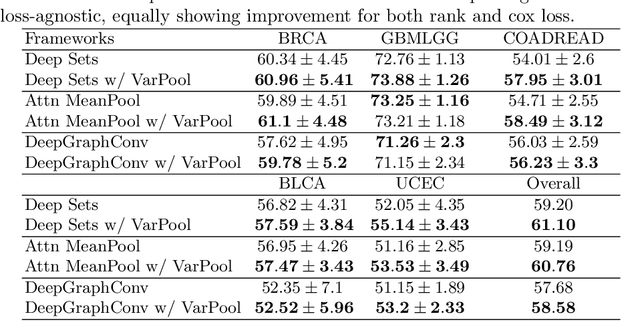
Supervised learning tasks such as cancer survival prediction from gigapixel whole slide images (WSIs) are a critical challenge in computational pathology that requires modeling complex features of the tumor microenvironment. These learning tasks are often solved with deep multi-instance learning (MIL) models that do not explicitly capture intratumoral heterogeneity. We develop a novel variance pooling architecture that enables a MIL model to incorporate intratumoral heterogeneity into its predictions. Two interpretability tools based on representative patches are illustrated to probe the biological signals captured by these models. An empirical study with 4,479 gigapixel WSIs from the Cancer Genome Atlas shows that adding variance pooling onto MIL frameworks improves survival prediction performance for five cancer types.
Scaling Vision Transformers to Gigapixel Images via Hierarchical Self-Supervised Learning
Jun 06, 2022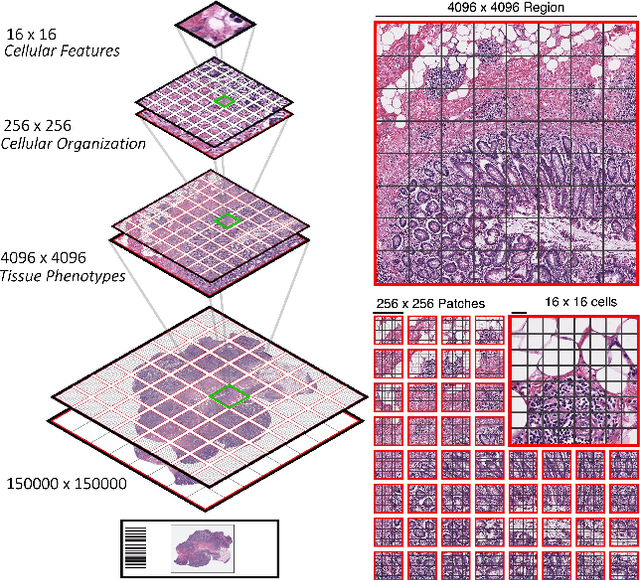
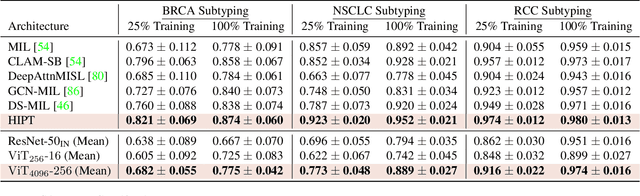
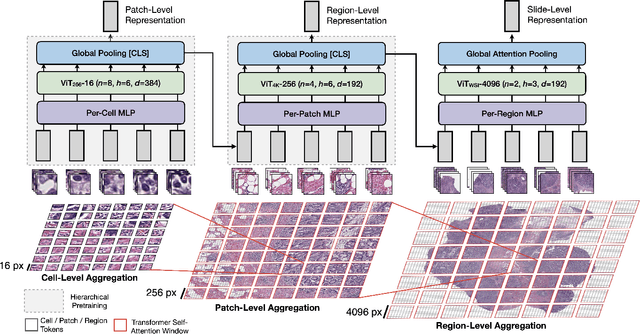

Vision Transformers (ViTs) and their multi-scale and hierarchical variations have been successful at capturing image representations but their use has been generally studied for low-resolution images (e.g. - 256x256, 384384). For gigapixel whole-slide imaging (WSI) in computational pathology, WSIs can be as large as 150000x150000 pixels at 20X magnification and exhibit a hierarchical structure of visual tokens across varying resolutions: from 16x16 images capture spatial patterns among cells, to 4096x4096 images characterizing interactions within the tissue microenvironment. We introduce a new ViT architecture called the Hierarchical Image Pyramid Transformer (HIPT), which leverages the natural hierarchical structure inherent in WSIs using two levels of self-supervised learning to learn high-resolution image representations. HIPT is pretrained across 33 cancer types using 10,678 gigapixel WSIs, 408,218 4096x4096 images, and 104M 256x256 images. We benchmark HIPT representations on 9 slide-level tasks, and demonstrate that: 1) HIPT with hierarchical pretraining outperforms current state-of-the-art methods for cancer subtyping and survival prediction, 2) self-supervised ViTs are able to model important inductive biases about the hierarchical structure of phenotypes in the tumor microenvironment.
Algorithm Fairness in AI for Medicine and Healthcare
Oct 01, 2021
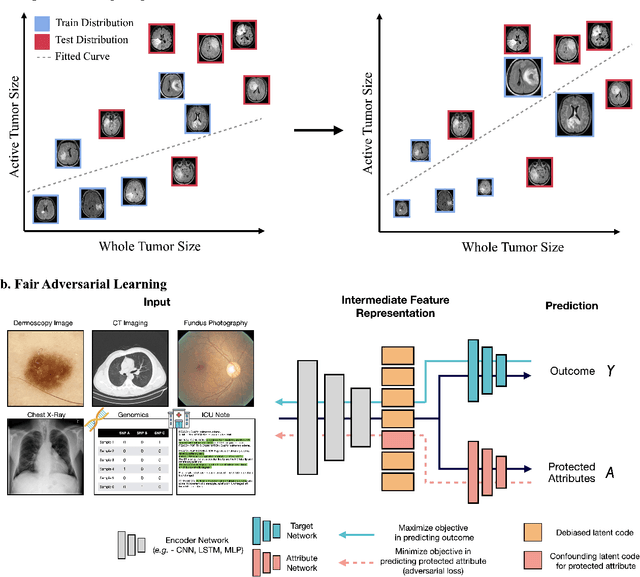

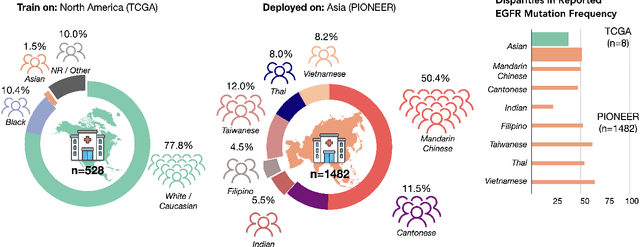
In the current development and deployment of many artificial intelligence (AI) systems in healthcare, algorithm fairness is a challenging problem in delivering equitable care. Recent evaluation of AI models stratified across race sub-populations have revealed enormous inequalities in how patients are diagnosed, given treatments, and billed for healthcare costs. In this perspective article, we summarize the intersectional field of fairness in machine learning through the context of current issues in healthcare, outline how algorithmic biases (e.g. - image acquisition, genetic variation, intra-observer labeling variability) arise in current clinical workflows and their resulting healthcare disparities. Lastly, we also review emerging strategies for mitigating bias via decentralized learning, disentanglement, and model explainability.
Pan-Cancer Integrative Histology-Genomic Analysis via Interpretable Multimodal Deep Learning
Aug 04, 2021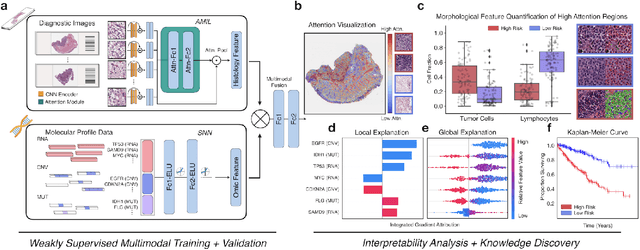
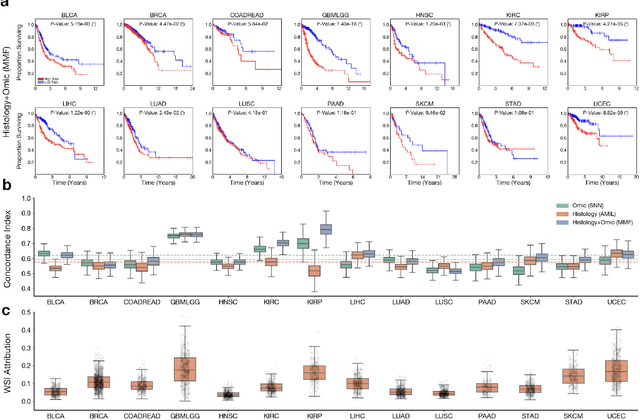
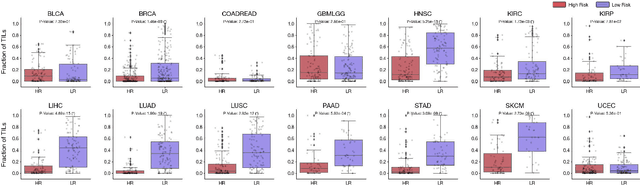
The rapidly emerging field of deep learning-based computational pathology has demonstrated promise in developing objective prognostic models from histology whole slide images. However, most prognostic models are either based on histology or genomics alone and do not address how histology and genomics can be integrated to develop joint image-omic prognostic models. Additionally identifying explainable morphological and molecular descriptors from these models that govern such prognosis is of interest. We used multimodal deep learning to integrate gigapixel whole slide pathology images, RNA-seq abundance, copy number variation, and mutation data from 5,720 patients across 14 major cancer types. Our interpretable, weakly-supervised, multimodal deep learning algorithm is able to fuse these heterogeneous modalities for predicting outcomes and discover prognostic features from these modalities that corroborate with poor and favorable outcomes via multimodal interpretability. We compared our model with unimodal deep learning models trained on histology slides and molecular profiles alone, and demonstrate performance increase in risk stratification on 9 out of 14 cancers. In addition, we analyze morphologic and molecular markers responsible for prognostic predictions across all cancer types. All analyzed data, including morphological and molecular correlates of patient prognosis across the 14 cancer types at a disease and patient level are presented in an interactive open-access database (http://pancancer.mahmoodlab.org) to allow for further exploration and prognostic biomarker discovery. To validate that these model explanations are prognostic, we further analyzed high attention morphological regions in WSIs, which indicates that tumor-infiltrating lymphocyte presence corroborates with favorable cancer prognosis on 9 out of 14 cancer types studied.
Fast and Scalable Image Search For Histology
Jul 28, 2021



The expanding adoption of digital pathology has enabled the curation of large repositories of histology whole slide images (WSIs), which contain a wealth of information. Similar pathology image search offers the opportunity to comb through large historical repositories of gigapixel WSIs to identify cases with similar morphological features and can be particularly useful for diagnosing rare diseases, identifying similar cases for predicting prognosis, treatment outcomes, and potential clinical trial success. A critical challenge in developing a WSI search and retrieval system is scalability, which is uniquely challenging given the need to search a growing number of slides that each can consist of billions of pixels and are several gigabytes in size. Such systems are typically slow and retrieval speed often scales with the size of the repository they search through, making their clinical adoption tedious and are not feasible for repositories that are constantly growing. Here we present Fast Image Search for Histopathology (FISH), a histology image search pipeline that is infinitely scalable and achieves constant search speed that is independent of the image database size while being interpretable and without requiring detailed annotations. FISH uses self-supervised deep learning to encode meaningful representations from WSIs and a Van Emde Boas tree for fast search, followed by an uncertainty-based ranking algorithm to retrieve similar WSIs. We evaluated FISH on multiple tasks and datasets with over 22,000 patient cases spanning 56 disease subtypes. We additionally demonstrate that FISH can be used to assist with the diagnosis of rare cancer types where sufficient cases may not be available to train traditional supervised deep models. FISH is available as an easy-to-use, open-source software package (https://github.com/mahmoodlab/FISH).
Deep Learning-based Frozen Section to FFPE Translation
Jul 27, 2021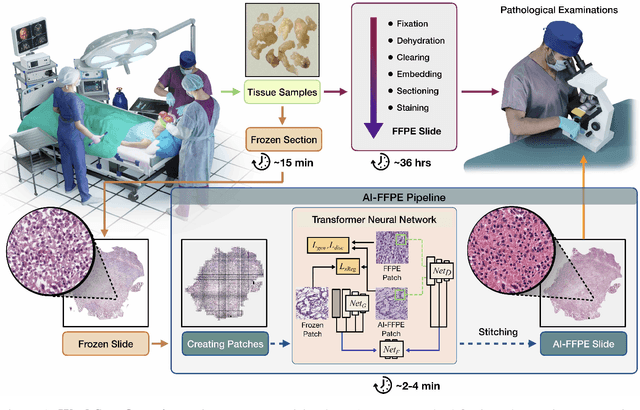
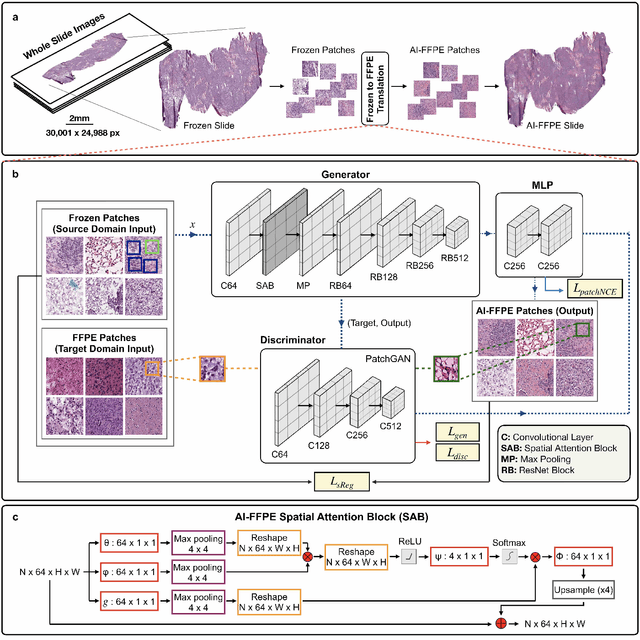
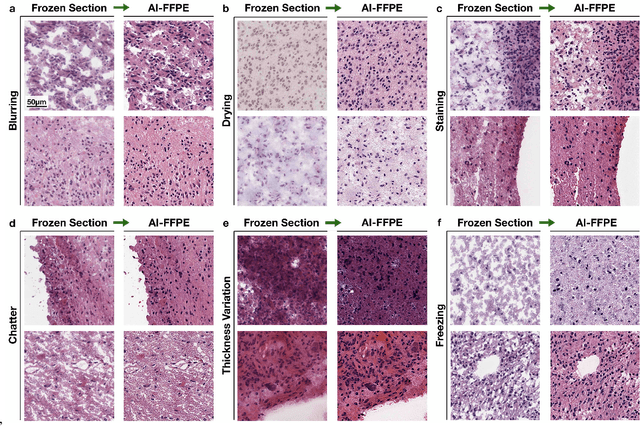
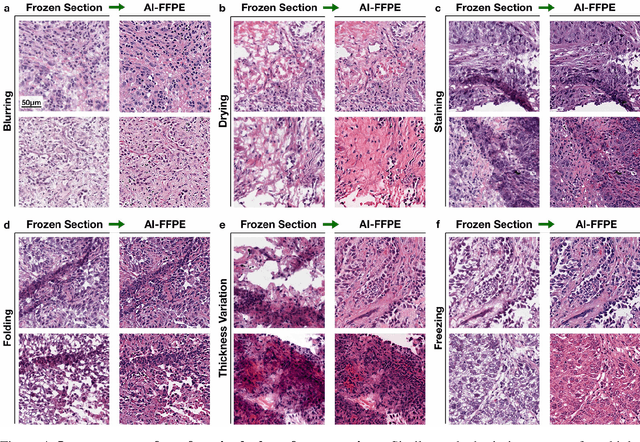
Frozen sectioning (FS) is the preparation method of choice for microscopic evaluation of tissues during surgical operations. The high speed of the procedure allows pathologists to rapidly assess the key microscopic features, such as tumour margins and malignant status to guide surgical decision-making and minimise disruptions to the course of the operation. However, FS is prone to introducing many misleading artificial structures (histological artefacts), such as nuclear ice crystals, compression, and cutting artefacts, hindering timely and accurate diagnostic judgement of the pathologist. Additional training and prolonged experience is often required to make highly effective and time-critical diagnosis on frozen sections. On the other hand, the gold standard tissue preparation technique of formalin-fixation and paraffin-embedding (FFPE) provides significantly superior image quality, but is a very time-consuming process (12-48 hours), making it unsuitable for intra-operative use. In this paper, we propose an artificial intelligence (AI) method that improves FS image quality by computationally transforming frozen-sectioned whole-slide images (FS-WSIs) into whole-slide FFPE-style images in minutes. AI-FFPE rectifies FS artefacts with the guidance of an attention mechanism that puts a particular emphasis on artefacts while utilising a self-regularization mechanism established between FS input image and synthesized FFPE-style image that preserves clinically relevant features. As a result, AI-FFPE method successfully generates FFPE-style images without significantly extending tissue processing time and consequently improves diagnostic accuracy. We demonstrate the efficacy of AI-FFPE on lung and brain frozen sections using a variety of different qualitative and quantitative metrics including visual Turing tests from 20 board certified pathologists.
Whole Slide Images are 2D Point Clouds: Context-Aware Survival Prediction using Patch-based Graph Convolutional Networks
Jul 27, 2021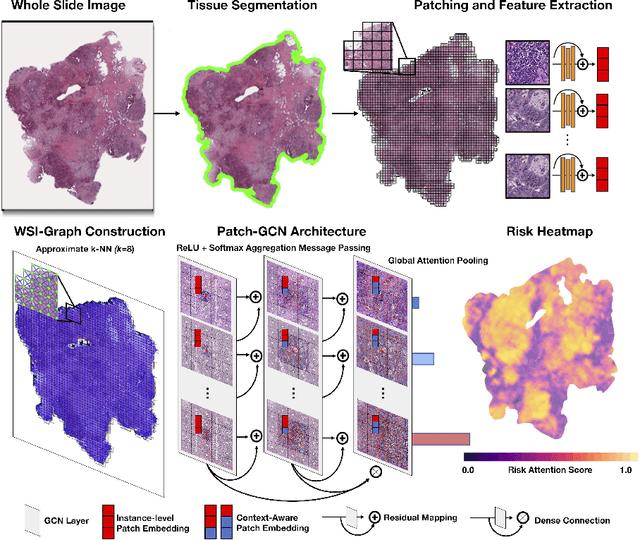
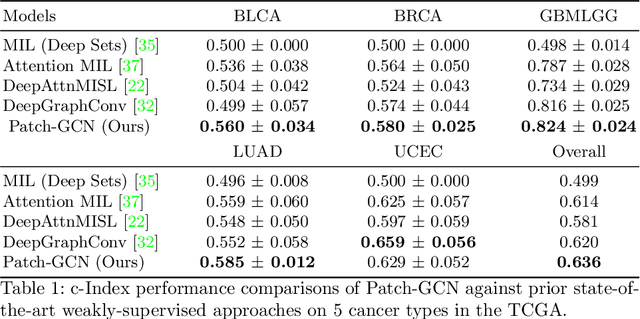
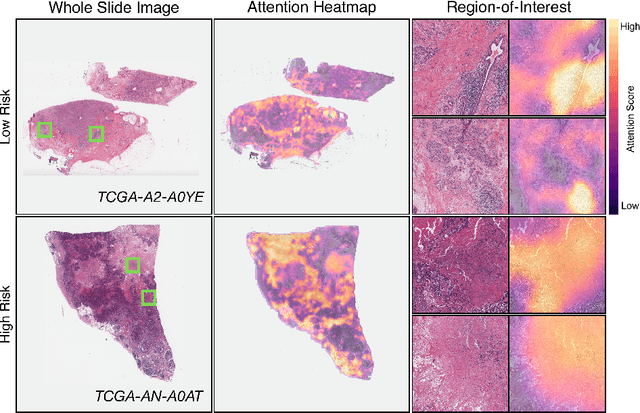
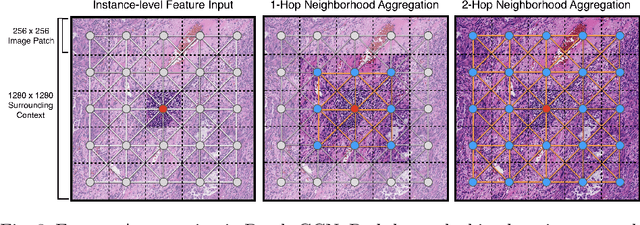
Cancer prognostication is a challenging task in computational pathology that requires context-aware representations of histology features to adequately infer patient survival. Despite the advancements made in weakly-supervised deep learning, many approaches are not context-aware and are unable to model important morphological feature interactions between cell identities and tissue types that are prognostic for patient survival. In this work, we present Patch-GCN, a context-aware, spatially-resolved patch-based graph convolutional network that hierarchically aggregates instance-level histology features to model local- and global-level topological structures in the tumor microenvironment. We validate Patch-GCN with 4,370 gigapixel WSIs across five different cancer types from the Cancer Genome Atlas (TCGA), and demonstrate that Patch-GCN outperforms all prior weakly-supervised approaches by 3.58-9.46%. Our code and corresponding models are publicly available at https://github.com/mahmoodlab/Patch-GCN.
Federated Learning for Computational Pathology on Gigapixel Whole Slide Images
Sep 23, 2020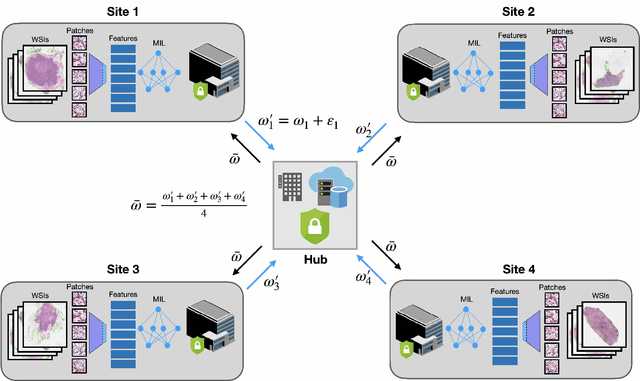
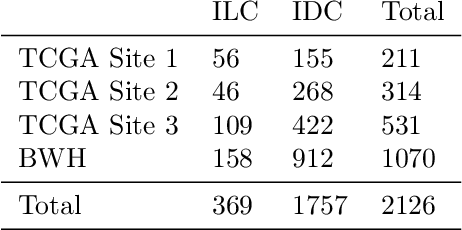
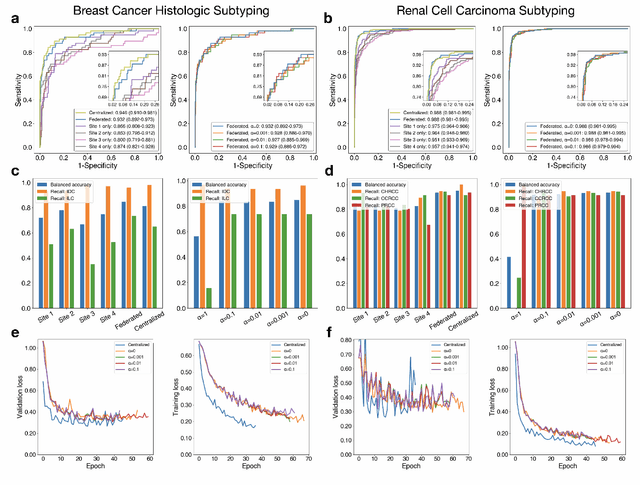
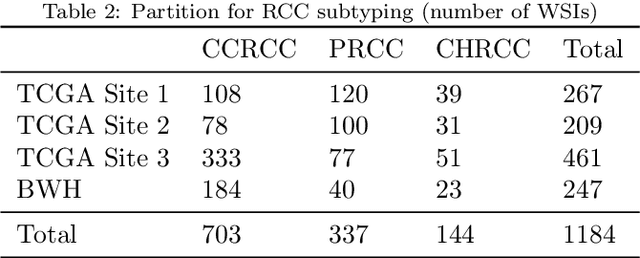
Deep Learning-based computational pathology algorithms have demonstrated profound ability to excel in a wide array of tasks that range from characterization of well known morphological phenotypes to predicting non-human-identifiable features from histology such as molecular alterations. However, the development of robust, adaptable, and accurate deep learning-based models often rely on the collection and time-costly curation large high-quality annotated training data that should ideally come from diverse sources and patient populations to cater for the heterogeneity that exists in such datasets. Multi-centric and collaborative integration of medical data across multiple institutions can naturally help overcome this challenge and boost the model performance but is limited by privacy concerns amongst other difficulties that may arise in the complex data sharing process as models scale towards using hundreds of thousands of gigapixel whole slide images. In this paper, we introduce privacy-preserving federated learning for gigapixel whole slide images in computational pathology using weakly-supervised attention multiple instance learning and differential privacy. We evaluated our approach on two different diagnostic problems using thousands of histology whole slide images with only slide-level labels. Additionally, we present a weakly-supervised learning framework for survival prediction and patient stratification from whole slide images and demonstrate its effectiveness in a federated setting. Our results show that using federated learning, we can effectively develop accurate weakly supervised deep learning models from distributed data silos without direct data sharing and its associated complexities, while also preserving differential privacy using randomized noise generation.
Deep Learning-based Computational Pathology Predicts Origins for Cancers of Unknown Primary
Jun 29, 2020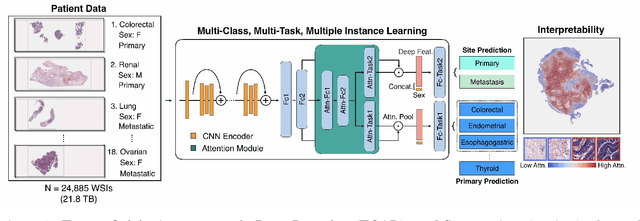
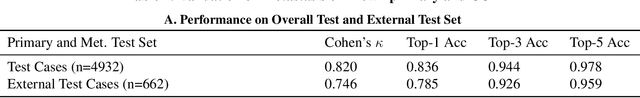
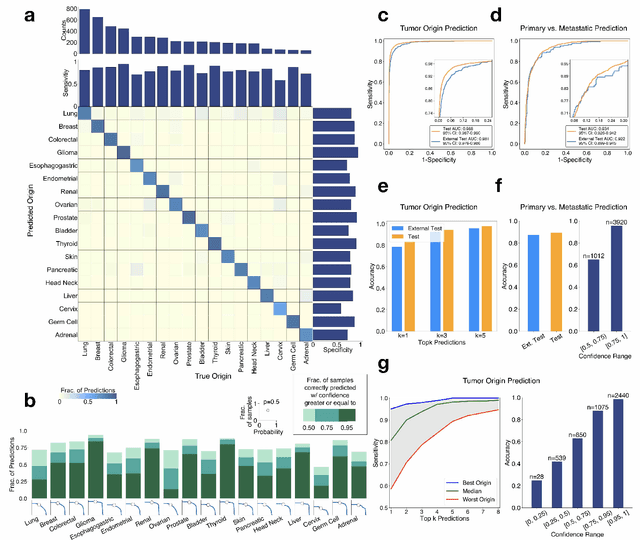
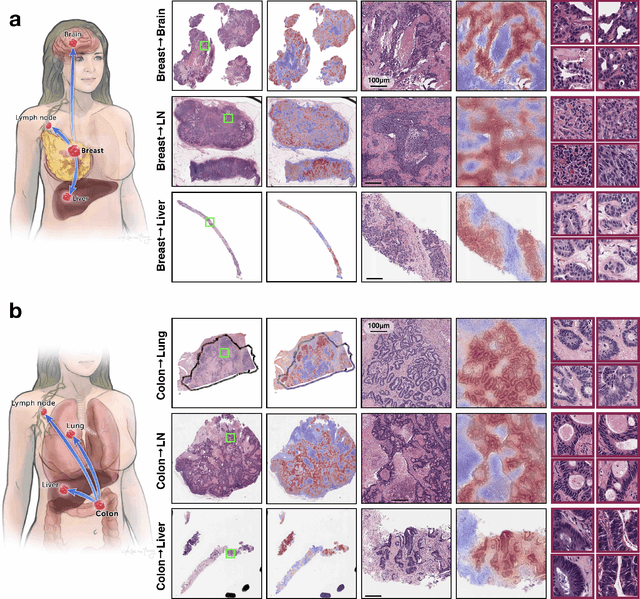
Cancer of unknown primary (CUP) is an enigmatic group of diagnoses where the primary anatomical site of tumor origin cannot be determined. This poses a significant challenge since modern therapeutics such as chemotherapy regimen and immune checkpoint inhibitors are specific to the primary tumor. Recent work has focused on using genomics and transcriptomics for identification of tumor origins. However, genomic testing is not conducted for every patient and lacks clinical penetration in low resource settings. Herein, to overcome these challenges, we present a deep learning-based computational pathology algorithm-TOAD-that can provide a differential diagnosis for CUP using routinely acquired histology slides. We used 17,486 gigapixel whole slide images with known primaries spread over 18 common origins to train a multi-task deep model to simultaneously identify the tumor as primary or metastatic and predict its site of origin. We tested our model on an internal test set of 4,932 cases with known primaries and achieved a top-1 accuracy of 0.84, a top-3 accuracy of 0.94 while on our external test set of 662 cases from 202 different hospitals, it achieved a top-1 and top-3 accuracy of 0.79 and 0.93 respectively. We further curated a dataset of 717 CUP cases from 151 different medical centers and identified a subset of 290 cases for which a differential diagnosis was assigned. Our model predictions resulted in concordance for 50% of cases (\k{appa}=0.4 when adjusted for agreement by chance) and a top-3 agreement of 75%. Our proposed method can be used as an assistive tool to assign differential diagnosis to complicated metastatic and CUP cases and could be used in conjunction with or in lieu of immunohistochemical analysis and extensive diagnostic work-ups to reduce the occurrence of CUP.
Data Efficient and Weakly Supervised Computational Pathology on Whole Slide Images
May 22, 2020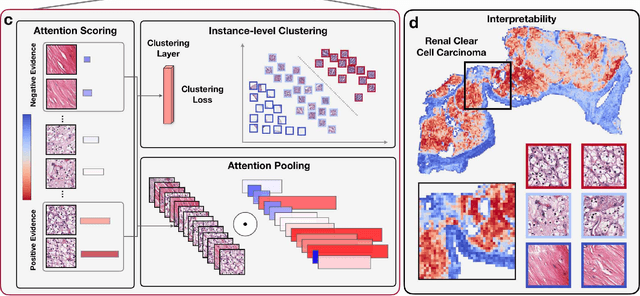
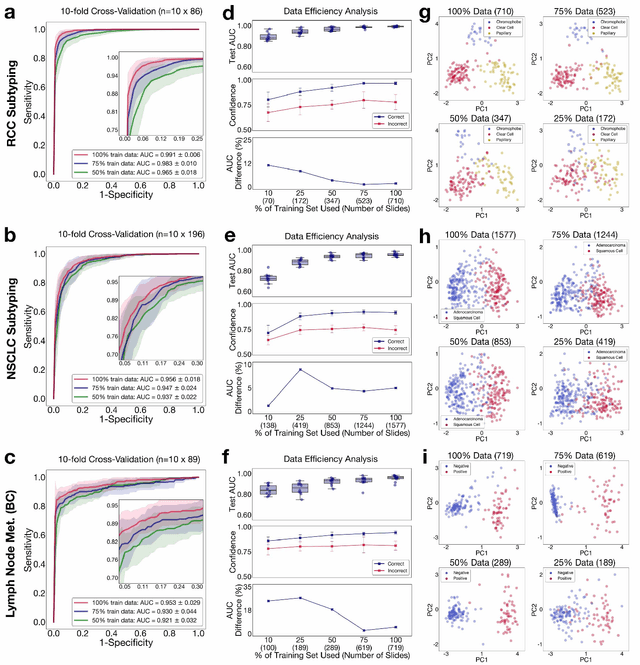
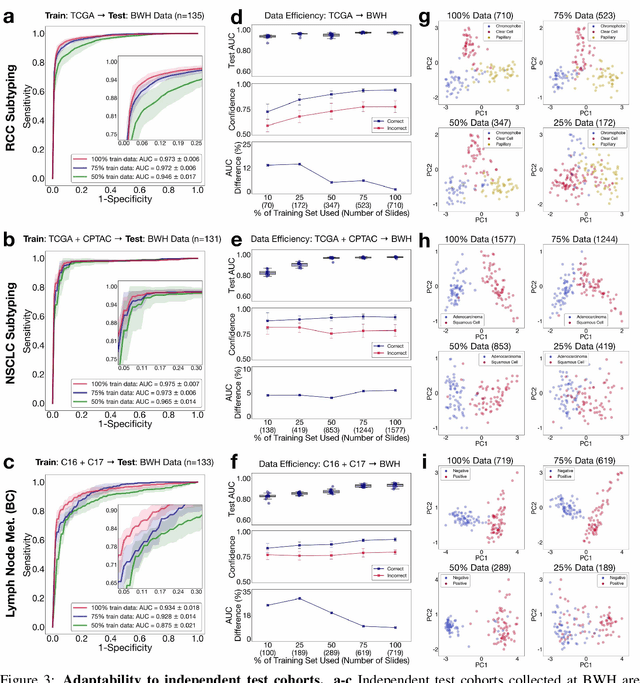
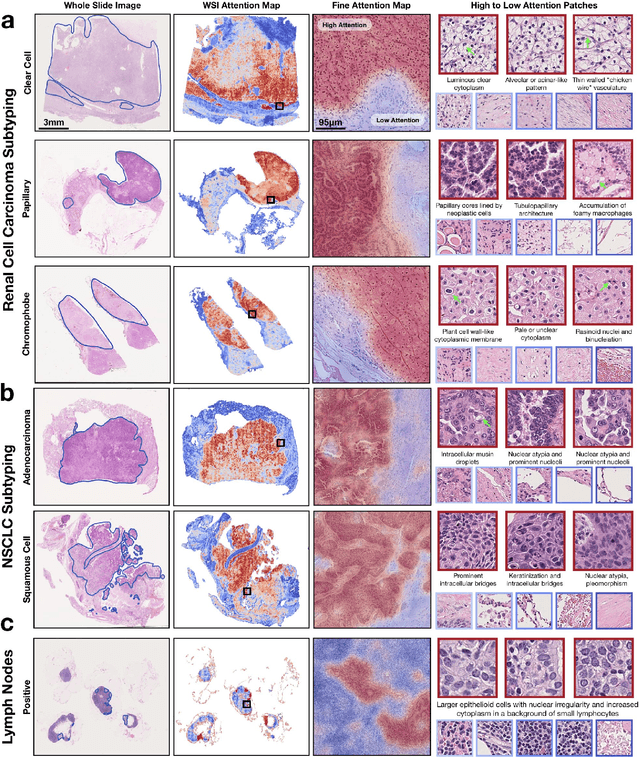
The rapidly emerging field of computational pathology has the potential to enable objective diagnosis, therapeutic response prediction and identification of new morphological features of clinical relevance. However, deep learning-based computational pathology approaches either require manual annotation of gigapixel whole slide images (WSIs) in fully-supervised settings or thousands of WSIs with slide-level labels in a weakly-supervised setting. Moreover, whole slide level computational pathology methods also suffer from domain adaptation and interpretability issues. These challenges have prevented the broad adaptation of computational pathology for clinical and research purposes. Here we present CLAM - Clustering-constrained attention multiple instance learning, an easy-to-use, high-throughput, and interpretable WSI-level processing and learning method that only requires slide-level labels while being data efficient, adaptable and capable of handling multi-class subtyping problems. CLAM is a deep-learning-based weakly-supervised method that uses attention-based learning to automatically identify sub-regions of high diagnostic value in order to accurately classify the whole slide, while also utilizing instance-level clustering over the representative regions identified to constrain and refine the feature space. In three separate analyses, we demonstrate the data efficiency and adaptability of CLAM and its superior performance over standard weakly-supervised classification. We demonstrate that CLAM models are interpretable and can be used to identify well-known and new morphological features. We further show that models trained using CLAM are adaptable to independent test cohorts, cell phone microscopy images, and biopsies. CLAM is a general-purpose and adaptable method that can be used for a variety of different computational pathology tasks in both clinical and research settings.