 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeometric Embedding Alignment via Curvature Matching in Transfer Learning
Jun 16, 2025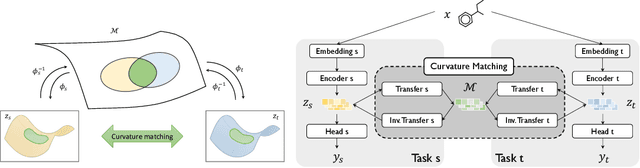

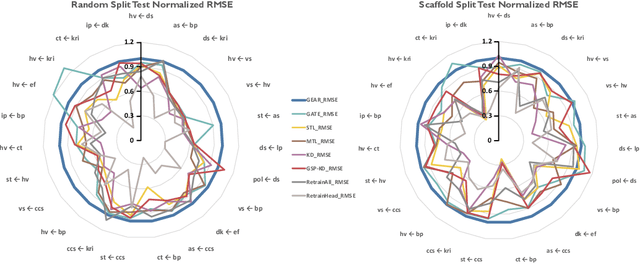

Geometrical interpretations of deep learning models offer insightful perspectives into their underlying mathematical structures. In this work, we introduce a novel approach that leverages differential geometry, particularly concepts from Riemannian geometry, to integrate multiple models into a unified transfer learning framework. By aligning the Ricci curvature of latent space of individual models, we construct an interrelated architecture, namely Geometric Embedding Alignment via cuRvature matching in transfer learning (GEAR), which ensures comprehensive geometric representation across datapoints. This framework enables the effective aggregation of knowledge from diverse sources, thereby improving performance on target tasks. We evaluate our model on 23 molecular task pairs sourced from various domains and demonstrate significant performance gains over existing benchmark model under both random (14.4%) and scaffold (8.3%) data splits.
Scalable Multi-Task Transfer Learning for Molecular Property Prediction
Oct 01, 2024



Molecules have a number of distinct properties whose importance and application vary. Often, in reality, labels for some properties are hard to achieve despite their practical importance. A common solution to such data scarcity is to use models of good generalization with transfer learning. This involves domain experts for designing source and target tasks whose features are shared. However, this approach has limitations: i). Difficulty in accurate design of source-target task pairs due to the large number of tasks, and ii). corresponding computational burden verifying many trials and errors of transfer learning design, thereby iii). constraining the potential of foundation modeling of multi-task molecular property prediction. We address the limitations of the manual design of transfer learning via data-driven bi-level optimization. The proposed method enables scalable multi-task transfer learning for molecular property prediction by automatically obtaining the optimal transfer ratios. Empirically, the proposed method improved the prediction performance of 40 molecular properties and accelerated training convergence.
Task Addition in Multi-Task Learning by Geometrical Alignment
Sep 25, 2024



Training deep learning models on limited data while maintaining generalization is one of the fundamental challenges in molecular property prediction. One effective solution is transferring knowledge extracted from abundant datasets to those with scarce data. Recently, a novel algorithm called Geometrically Aligned Transfer Encoder (GATE) has been introduced, which uses soft parameter sharing by aligning the geometrical shapes of task-specific latent spaces. However, GATE faces limitations in scaling to multiple tasks due to computational costs. In this study, we propose a task addition approach for GATE to improve performance on target tasks with limited data while minimizing computational complexity. It is achieved through supervised multi-task pre-training on a large dataset, followed by the addition and training of task-specific modules for each target task. Our experiments demonstrate the superior performance of the task addition strategy for GATE over conventional multi-task methods, with comparable computational costs.
Multitask Extension of Geometrically Aligned Transfer Encoder
May 03, 2024



Molecular datasets often suffer from a lack of data. It is well-known that gathering data is difficult due to the complexity of experimentation or simulation involved. Here, we leverage mutual information across different tasks in molecular data to address this issue. We extend an algorithm that utilizes the geometric characteristics of the encoding space, known as the Geometrically Aligned Transfer Encoder (GATE), to a multi-task setup. Thus, we connect multiple molecular tasks by aligning the curved coordinates onto locally flat coordinates, ensuring the flow of information from source tasks to support performance on target data.
Geometrically Aligned Transfer Encoder for Inductive Transfer in Regression Tasks
Oct 10, 2023
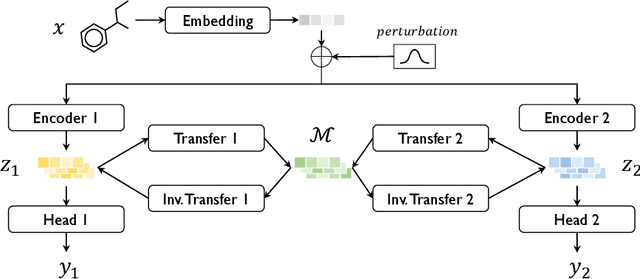
Transfer learning is a crucial technique for handling a small amount of data that is potentially related to other abundant data. However, most of the existing methods are focused on classification tasks using images and language datasets. Therefore, in order to expand the transfer learning scheme to regression tasks, we propose a novel transfer technique based on differential geometry, namely the Geometrically Aligned Transfer Encoder (GATE). In this method, we interpret the latent vectors from the model to exist on a Riemannian curved manifold. We find a proper diffeomorphism between pairs of tasks to ensure that every arbitrary point maps to a locally flat coordinate in the overlapping region, allowing the transfer of knowledge from the source to the target data. This also serves as an effective regularizer for the model to behave in extrapolation regions. In this article, we demonstrate that GATE outperforms conventional methods and exhibits stable behavior in both the latent space and extrapolation regions for various molecular graph datasets.
3D Denoisers are Good 2D Teachers: Molecular Pretraining via Denoising and Cross-Modal Distillation
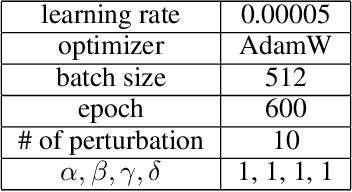
Sep 08, 2023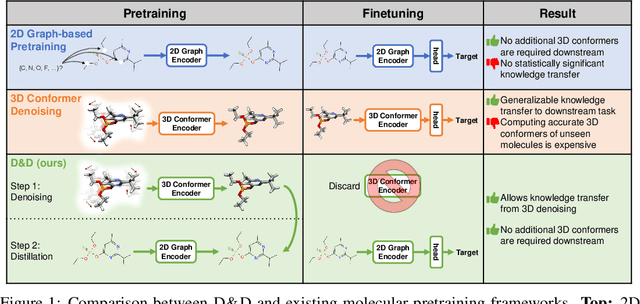
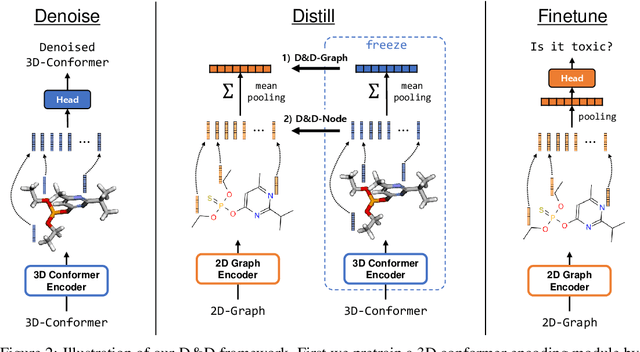
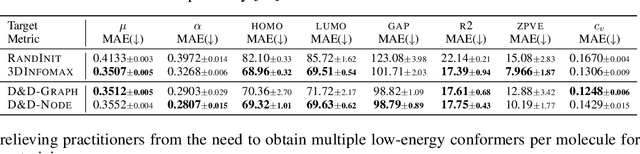
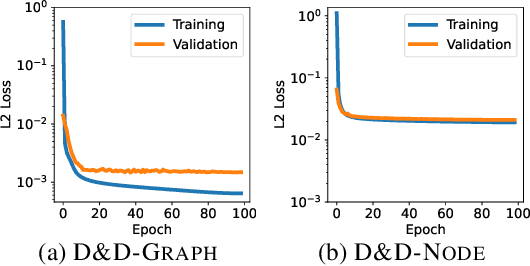
Pretraining molecular representations from large unlabeled data is essential for molecular property prediction due to the high cost of obtaining ground-truth labels. While there exist various 2D graph-based molecular pretraining approaches, these methods struggle to show statistically significant gains in predictive performance. Recent work have thus instead proposed 3D conformer-based pretraining under the task of denoising, which led to promising results. During downstream finetuning, however, models trained with 3D conformers require accurate atom-coordinates of previously unseen molecules, which are computationally expensive to acquire at scale. In light of this limitation, we propose D&D, a self-supervised molecular representation learning framework that pretrains a 2D graph encoder by distilling representations from a 3D denoiser. With denoising followed by cross-modal knowledge distillation, our approach enjoys use of knowledge obtained from denoising as well as painless application to downstream tasks with no access to accurate conformers. Experiments on real-world molecular property prediction datasets show that the graph encoder trained via D&D can infer 3D information based on the 2D graph and shows superior performance and label-efficiency against other baselines.
Grouping-matrix based Graph Pooling with Adaptive Number of Clusters
Sep 07, 2022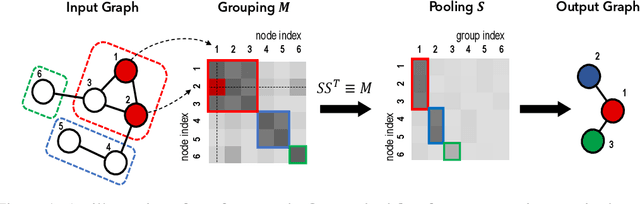
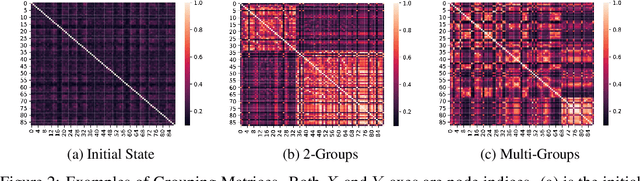
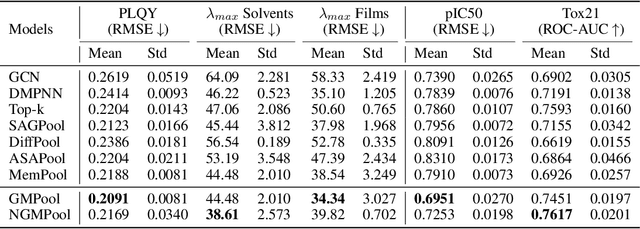
Graph pooling is a crucial operation for encoding hierarchical structures within graphs. Most existing graph pooling approaches formulate the problem as a node clustering task which effectively captures the graph topology. Conventional methods ask users to specify an appropriate number of clusters as a hyperparameter, then assume that all input graphs share the same number of clusters. In inductive settings where the number of clusters can vary, however, the model should be able to represent this variation in its pooling layers in order to learn suitable clusters. Thus we propose GMPool, a novel differentiable graph pooling architecture that automatically determines the appropriate number of clusters based on the input data. The main intuition involves a grouping matrix defined as a quadratic form of the pooling operator, which induces use of binary classification probabilities of pairwise combinations of nodes. GMPool obtains the pooling operator by first computing the grouping matrix, then decomposing it. Extensive evaluations on molecular property prediction tasks demonstrate that our method outperforms conventional methods.