 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInverse Lithography Physics-informed Deep Neural Level Set for Mask Optimization
Aug 15, 2023As the feature size of integrated circuits continues to decrease, optical proximity correction (OPC) has emerged as a crucial resolution enhancement technology for ensuring high printability in the lithography process. Recently, level set-based inverse lithography technology (ILT) has drawn considerable attention as a promising OPC solution, showcasing its powerful pattern fidelity, especially in advanced process. However, massive computational time consumption of ILT limits its applicability to mainly correcting partial layers and hotspot regions. Deep learning (DL) methods have shown great potential in accelerating ILT. However, lack of domain knowledge of inverse lithography limits the ability of DL-based algorithms in process window (PW) enhancement and etc. In this paper, we propose an inverse lithography physics-informed deep neural level set (ILDLS) approach for mask optimization. This approach utilizes level set based-ILT as a layer within the DL framework and iteratively conducts mask prediction and correction to significantly enhance printability and PW in comparison with results from pure DL and ILT. With this approach, computation time is reduced by a few orders of magnitude versus ILT. By gearing up DL with knowledge of inverse lithography physics, ILDLS provides a new and efficient mask optimization solution.
DR-Label: Improving GNN Models for Catalysis Systems by Label Deconstruction and Reconstruction
Mar 06, 2023Attaining the equilibrium state of a catalyst-adsorbate system is key to fundamentally assessing its effective properties, such as adsorption energy. Machine learning methods with finer supervision strategies have been applied to boost and guide the relaxation process of an atomic system and better predict its properties at the equilibrium state. In this paper, we present a novel graph neural network (GNN) supervision and prediction strategy DR-Label. The method enhances the supervision signal, reduces the multiplicity of solutions in edge representation, and encourages the model to provide node predictions that are graph structural variation robust. DR-Label first Deconstructs finer-grained equilibrium state information to the model by projecting the node-level supervision signal to each edge. Reversely, the model Reconstructs a more robust equilibrium state prediction by transforming edge-level predictions to node-level with a sphere-fitting algorithm. The DR-Label strategy was applied to three radically distinct models, each of which displayed consistent performance enhancements. Based on the DR-Label strategy, we further proposed DRFormer, which achieved a new state-of-the-art performance on the Open Catalyst 2020 (OC20) dataset and the Cu-based single-atom-alloyed CO adsorption (SAA) dataset. We expect that our work will highlight crucial steps for the development of a more accurate model in equilibrium state property prediction of a catalysis system.
Multi-Task Mixture Density Graph Neural Networks for Predicting Cu-based Single-Atom Alloy Catalysts for CO2 Reduction Reaction
Sep 15, 2022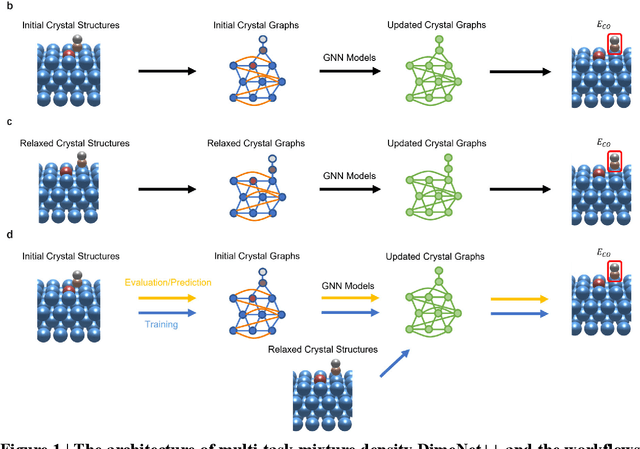
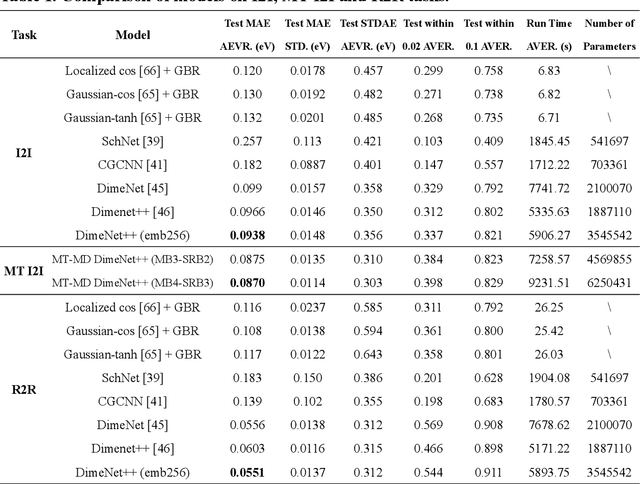
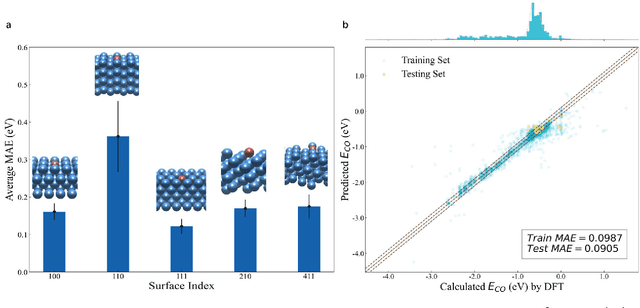
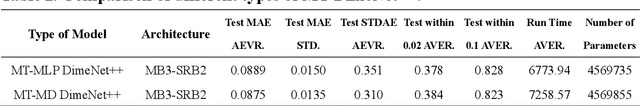
Graph neural networks (GNNs) have drawn more and more attention from material scientists and demonstrated a high capacity to establish connections between the structure and properties. However, with only unrelaxed structures provided as input, few GNN models can predict the thermodynamic properties of relaxed configurations with an acceptable level of error. In this work, we develop a multi-task (MT) architecture based on DimeNet++ and mixture density networks to improve the performance of such task. Taking CO adsorption on Cu-based single-atom alloy catalysts as an illustration, we show that our method can reliably estimate CO adsorption energy with a mean absolute error of 0.087 eV from the initial CO adsorption structures without costly first-principles calculations. Further, compared to other state-of-the-art GNN methods, our model exhibits improved generalization ability when predicting catalytic performance of out-of-domain configurations, built with either unseen substrate surfaces or doping species. We show that the proposed MT GNN strategy can facilitate catalyst discovery.
Heterogeneous relational message passing networks for molecular dynamics simulations
Sep 02, 2021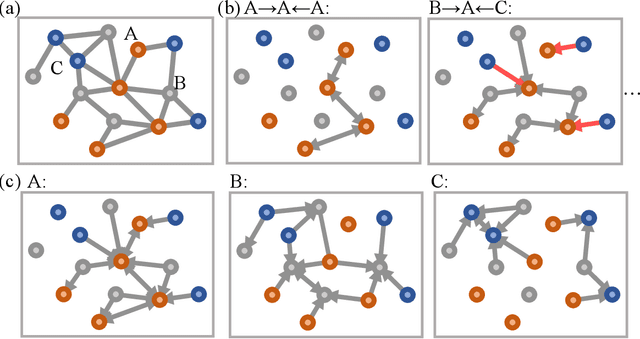
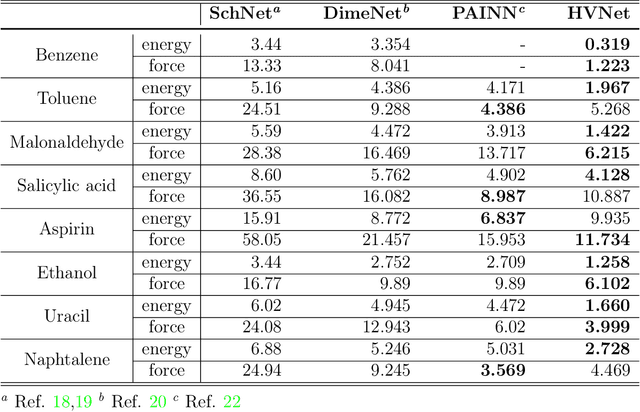
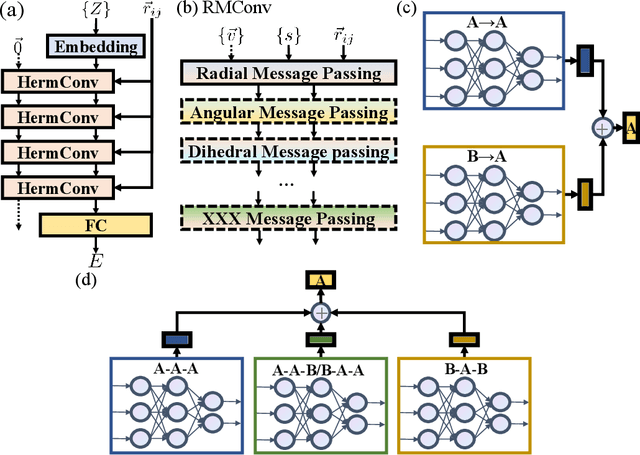
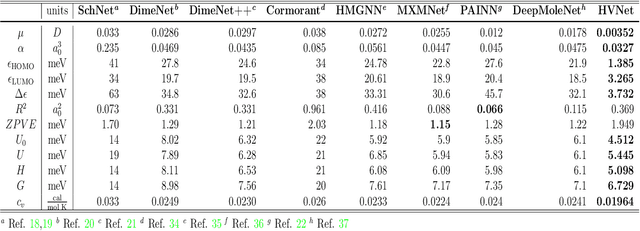
With many frameworks based on message passing neural networks proposed to predict molecular and bulk properties, machine learning methods have tremendously shifted the paradigms of computational sciences underpinning physics, material science, chemistry, and biology. While existing machine learning models have yielded superior performances in many occasions, most of them model and process molecular systems in terms of homogeneous graph, which severely limits the expressive power for representing diverse interactions. In practice, graph data with multiple node and edge types is ubiquitous and more appropriate for molecular systems. Thus, we propose the heterogeneous relational message passing network (HermNet), an end-to-end heterogeneous graph neural networks, to efficiently express multiple interactions in a single model with {\it ab initio} accuracy. HermNet performs impressively against many top-performing models on both molecular and extended systems. Specifically, HermNet outperforms other tested models in nearly 75\%, 83\% and 94\% of tasks on MD17, QM9 and extended systems datasets, respectively. Finally, we elucidate how the design of HermNet is compatible with quantum mechanics from the perspective of the density functional theory. Besides, HermNet is a universal framework, whose sub-networks could be replaced by other advanced models.