 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-Task Mixture Density Graph Neural Networks for Predicting Cu-based Single-Atom Alloy Catalysts for CO2 Reduction Reaction
Paper and Code
Sep 15, 2022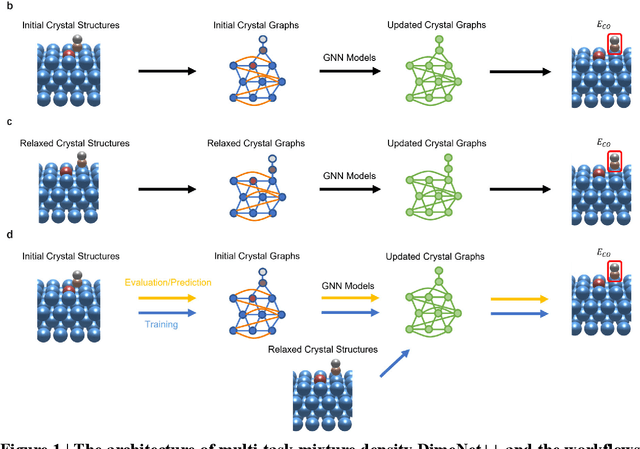
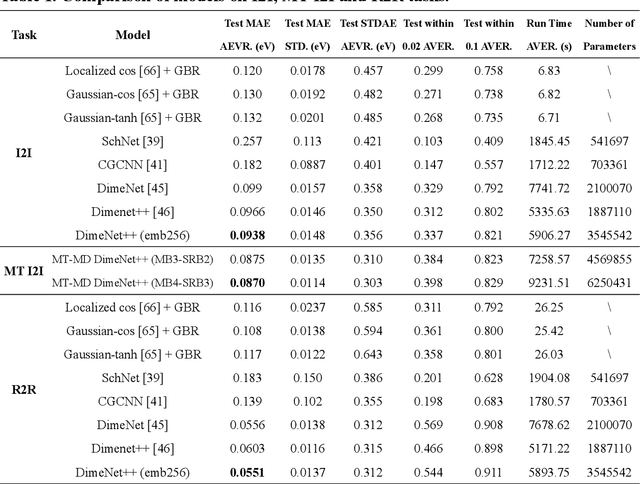
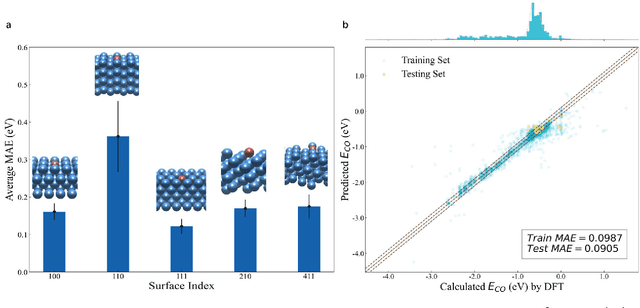
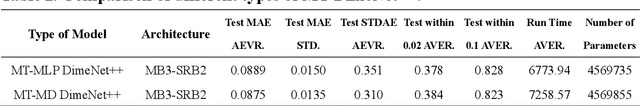
Graph neural networks (GNNs) have drawn more and more attention from material scientists and demonstrated a high capacity to establish connections between the structure and properties. However, with only unrelaxed structures provided as input, few GNN models can predict the thermodynamic properties of relaxed configurations with an acceptable level of error. In this work, we develop a multi-task (MT) architecture based on DimeNet++ and mixture density networks to improve the performance of such task. Taking CO adsorption on Cu-based single-atom alloy catalysts as an illustration, we show that our method can reliably estimate CO adsorption energy with a mean absolute error of 0.087 eV from the initial CO adsorption structures without costly first-principles calculations. Further, compared to other state-of-the-art GNN methods, our model exhibits improved generalization ability when predicting catalytic performance of out-of-domain configurations, built with either unseen substrate surfaces or doping species. We show that the proposed MT GNN strategy can facilitate catalyst discovery.