 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRule-Assisted Attribute Embedding
Jun 10, 2025Recommendation systems often overlook the rich attribute information embedded in property graphs, limiting their effectiveness. Existing graph convolutional network (GCN) models either ignore attributes or rely on simplistic <user, item, attribute> triples, failing to capture deeper semantic structures. We propose RAE (Rule- Assisted Approach for Attribute Embedding), a novel method that improves recommendations by mining semantic rules from property graphs to guide attribute embedding. RAE performs rule-based random walks to generate enriched attribute representations, which are integrated into GCNs. Experiments on real-world datasets (BlogCatalog, Flickr) show that RAE outperforms state-of-the-art baselines by 10.6% on average in Recall@20 and NDCG@20. RAE also demonstrates greater robustness to sparse data and missing attributes, highlighting the value of leveraging structured attribute information in recommendation tasks.
Heterogeneous relational message passing networks for molecular dynamics simulations
Sep 02, 2021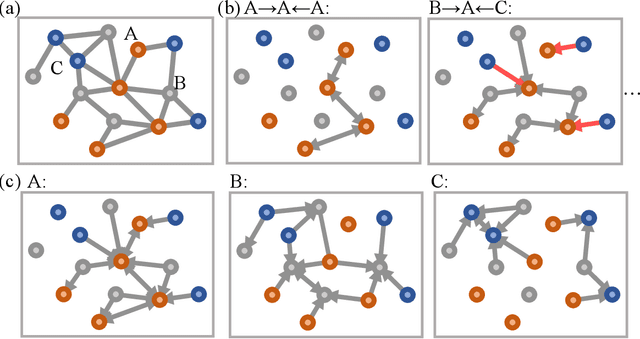
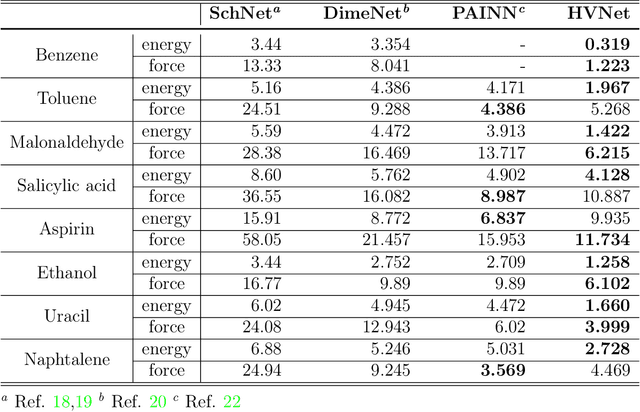
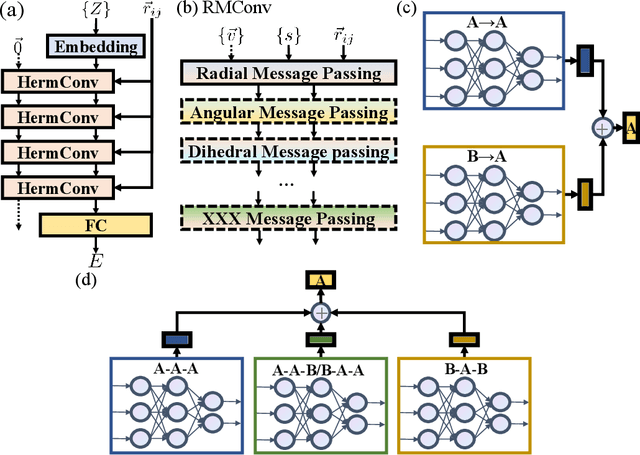
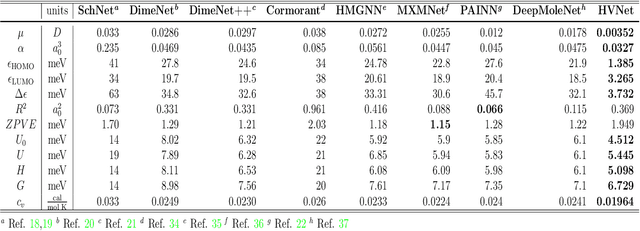
With many frameworks based on message passing neural networks proposed to predict molecular and bulk properties, machine learning methods have tremendously shifted the paradigms of computational sciences underpinning physics, material science, chemistry, and biology. While existing machine learning models have yielded superior performances in many occasions, most of them model and process molecular systems in terms of homogeneous graph, which severely limits the expressive power for representing diverse interactions. In practice, graph data with multiple node and edge types is ubiquitous and more appropriate for molecular systems. Thus, we propose the heterogeneous relational message passing network (HermNet), an end-to-end heterogeneous graph neural networks, to efficiently express multiple interactions in a single model with {\it ab initio} accuracy. HermNet performs impressively against many top-performing models on both molecular and extended systems. Specifically, HermNet outperforms other tested models in nearly 75\%, 83\% and 94\% of tasks on MD17, QM9 and extended systems datasets, respectively. Finally, we elucidate how the design of HermNet is compatible with quantum mechanics from the perspective of the density functional theory. Besides, HermNet is a universal framework, whose sub-networks could be replaced by other advanced models.
Symmetry-adapted graph neural networks for constructing molecular dynamics force fields
Jan 08, 2021



Molecular dynamics is a powerful simulation tool to explore material properties. Most of the realistic material systems are too large to be simulated with first-principles molecular dynamics. Classical molecular dynamics has lower computational cost but requires accurate force fields to achieve chemical accuracy. In this work, we develop a symmetry-adapted graph neural networks framework, named molecular dynamics graph neural networks (MDGNN), to construct force fields automatically for molecular dynamics simulations for both molecules and crystals. This architecture consistently preserves the translation, rotation and permutation invariance in the simulations. We propose a new feature engineering method including higher order contributions and show that MDGNN accurately reproduces the results of both classical and first-principles molecular dynamics. We also demonstrate that force fields constructed by the model has good transferability. Therefore, MDGNN provides an efficient and promising option for molecular dynamics simulations of large scale systems with high accuracy.