 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOvercoming Practical Issues of Deep Active Learning and its Applications on Named Entity Recognition
Nov 17, 2019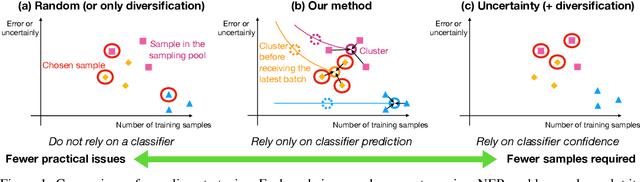
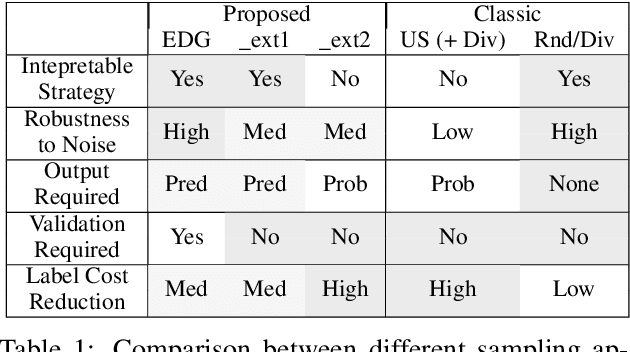
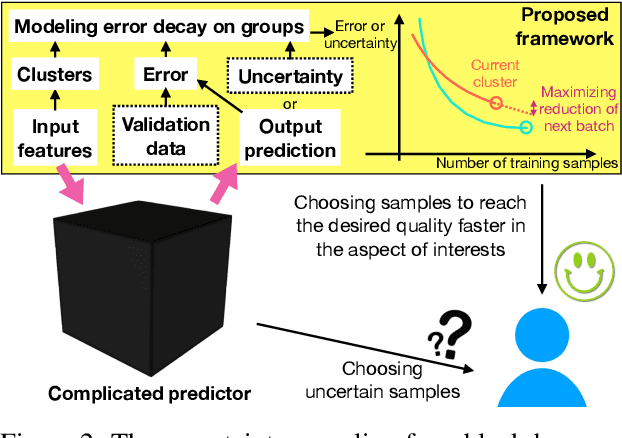
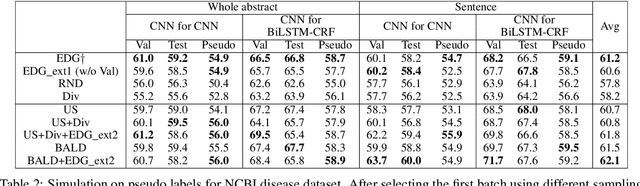
Existing deep active learning algorithms achieve impressive sampling efficiency on natural language processing tasks. However, they exhibit several weaknesses in practice, including (a) inability to use uncertainty sampling with black-box models, (b) lack of robustness to noise in labeling, (c) lack of transparency. In response, we propose a transparent batch active sampling framework by estimating the error decay curves of multiple feature-defined subsets of the data. Experiments on four named entity recognition (NER) tasks demonstrate that the proposed methods significantly outperform diversification-based methods for black-box NER taggers and can make the sampling process more robust to labeling noise when combined with uncertainty-based methods. Furthermore, the analysis of experimental results sheds light on the weaknesses of different active sampling strategies, and when traditional uncertainty-based or diversification-based methods can be expected to work well.
Interactive Learning from Multiple Noisy Labels
Jul 24, 2016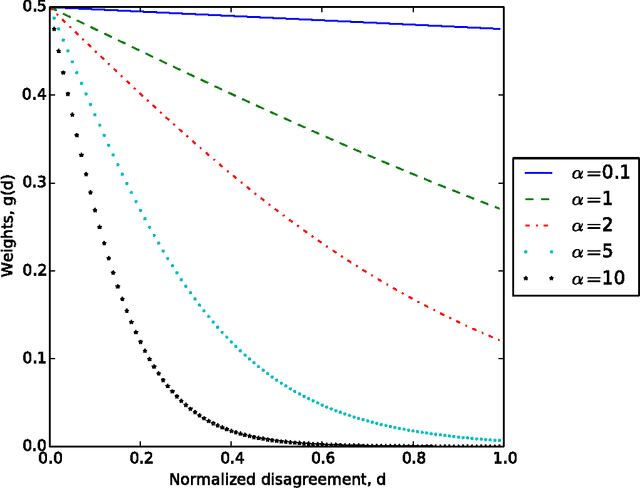
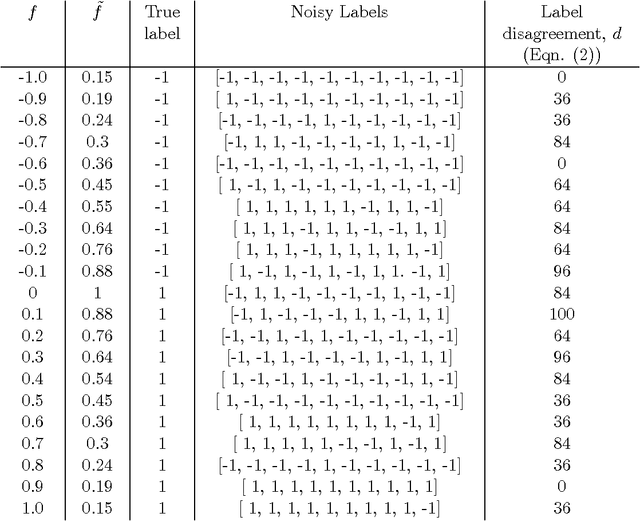
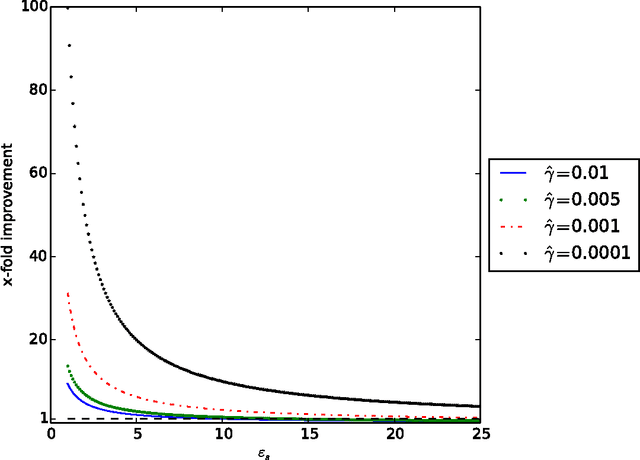
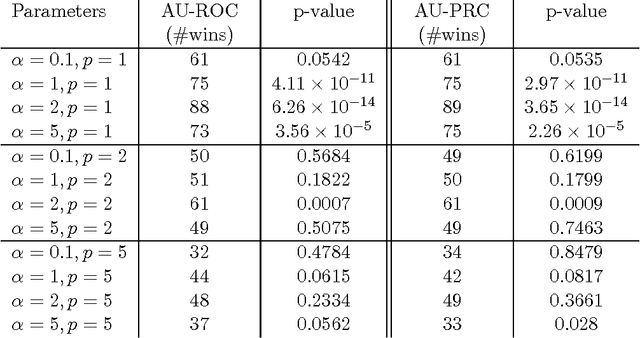
Interactive learning is a process in which a machine learning algorithm is provided with meaningful, well-chosen examples as opposed to randomly chosen examples typical in standard supervised learning. In this paper, we propose a new method for interactive learning from multiple noisy labels where we exploit the disagreement among annotators to quantify the easiness (or meaningfulness) of an example. We demonstrate the usefulness of this method in estimating the parameters of a latent variable classification model, and conduct experimental analyses on a range of synthetic and benchmark datasets. Furthermore, we theoretically analyze the performance of perceptron in this interactive learning framework.
Reconstructing subclonal composition and evolution from whole genome sequencing of tumors
Jan 06, 2015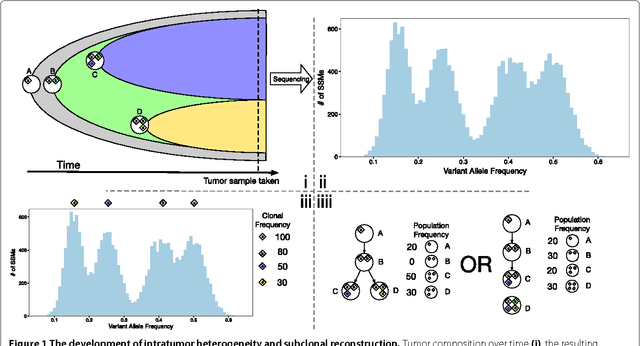
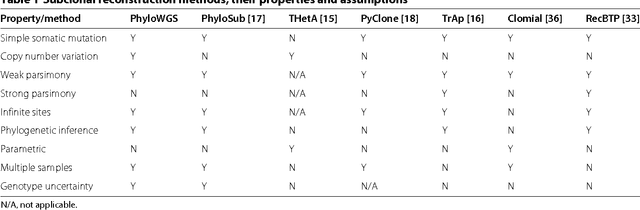
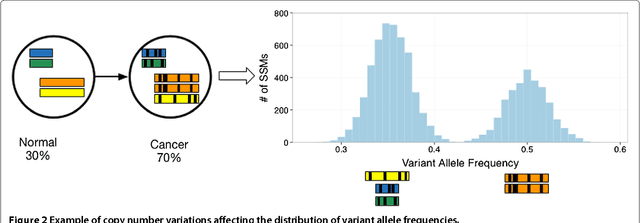
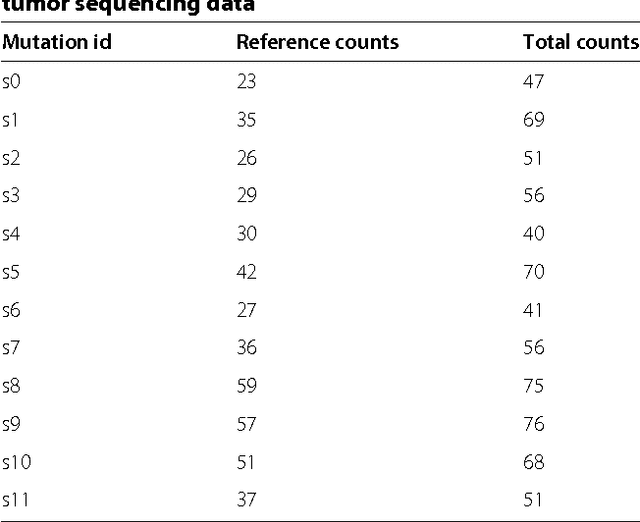
Tumors often contain multiple subpopulations of cancerous cells defined by distinct somatic mutations. We describe a new method, PhyloWGS, that can be applied to WGS data from one or more tumor samples to reconstruct complete genotypes of these subpopulations based on variant allele frequencies (VAFs) of point mutations and population frequencies of structural variations. We introduce a principled phylogenic correction for VAFs in loci affected by copy number alterations and we show that this correction greatly improves subclonal reconstruction compared to existing methods.
Comparing Nonparametric Bayesian Tree Priors for Clonal Reconstruction of Tumors
Aug 11, 2014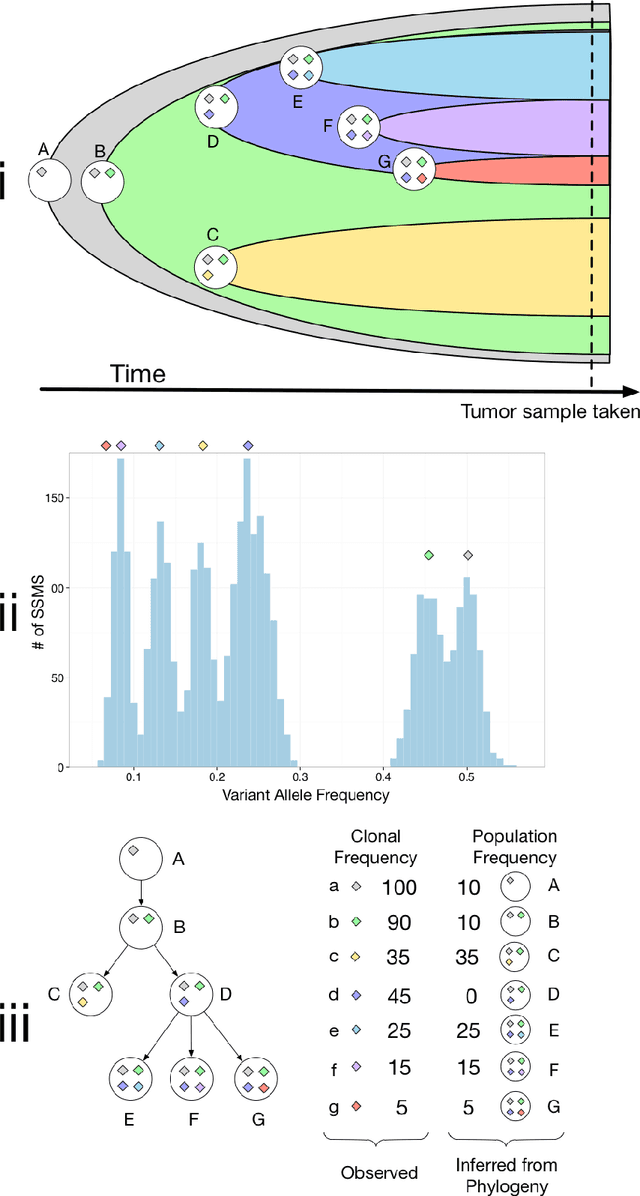
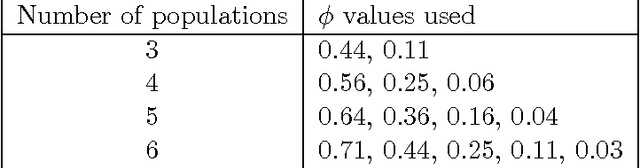
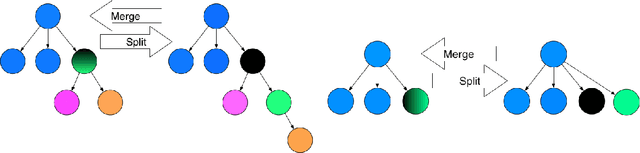
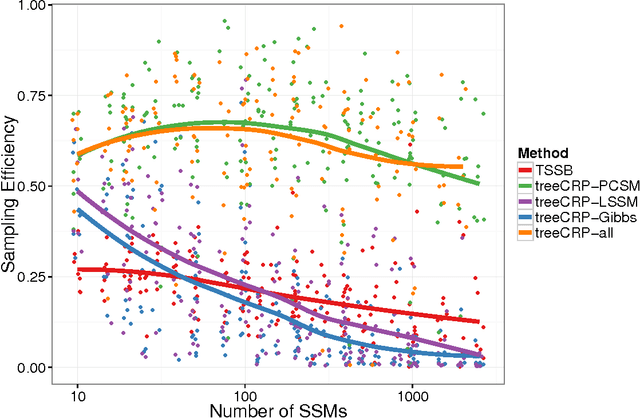
Statistical machine learning methods, especially nonparametric Bayesian methods, have become increasingly popular to infer clonal population structure of tumors. Here we describe the treeCRP, an extension of the Chinese restaurant process (CRP), a popular construction used in nonparametric mixture models, to infer the phylogeny and genotype of major subclonal lineages represented in the population of cancer cells. We also propose new split-merge updates tailored to the subclonal reconstruction problem that improve the mixing time of Markov chains. In comparisons with the tree-structured stick breaking prior used in PhyloSub, we demonstrate superior mixing and running time using the treeCRP with our new split-merge procedures. We also show that given the same number of samples, TSSB and treeCRP have similar ability to recover the subclonal structure of a tumor.
Inferring clonal evolution of tumors from single nucleotide somatic mutations
Nov 02, 2013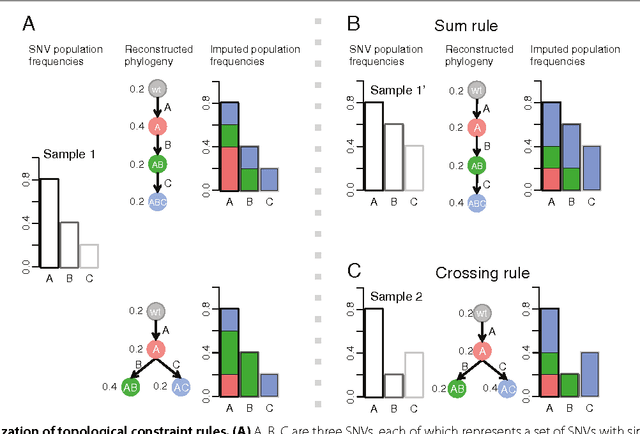
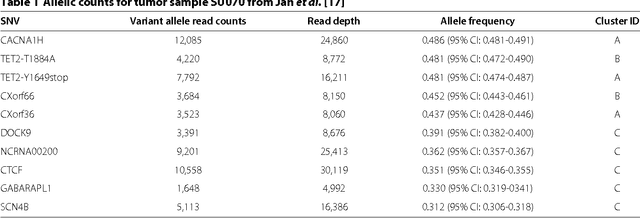
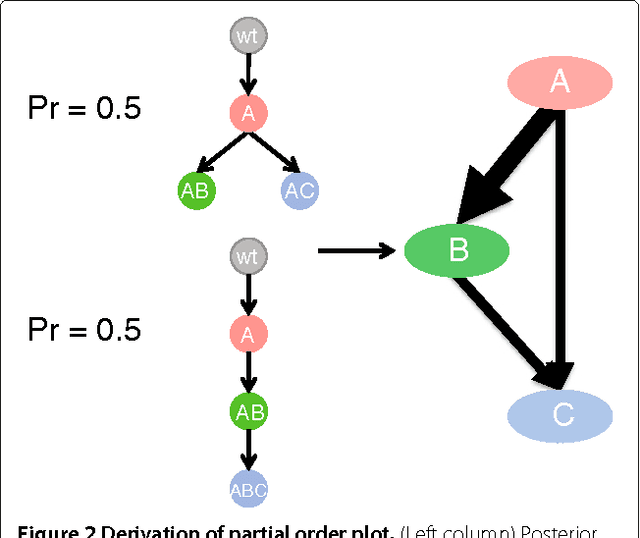
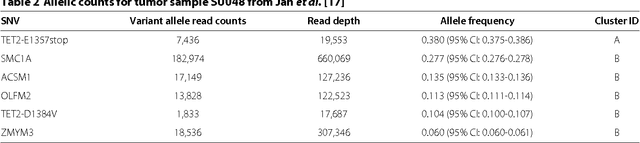
High-throughput sequencing allows the detection and quantification of frequencies of somatic single nucleotide variants (SNV) in heterogeneous tumor cell populations. In some cases, the evolutionary history and population frequency of the subclonal lineages of tumor cells present in the sample can be reconstructed from these SNV frequency measurements. However, automated methods to do this reconstruction are not available and the conditions under which reconstruction is possible have not been described. We describe the conditions under which the evolutionary history can be uniquely reconstructed from SNV frequencies from single or multiple samples from the tumor population and we introduce a new statistical model, PhyloSub, that infers the phylogeny and genotype of the major subclonal lineages represented in the population of cancer cells. It uses a Bayesian nonparametric prior over trees that groups SNVs into major subclonal lineages and automatically estimates the number of lineages and their ancestry. We sample from the joint posterior distribution over trees to identify evolutionary histories and cell population frequencies that have the highest probability of generating the observed SNV frequency data. When multiple phylogenies are consistent with a given set of SNV frequencies, PhyloSub represents the uncertainty in the tumor phylogeny using a partial order plot. Experiments on a simulated dataset and two real datasets comprising tumor samples from acute myeloid leukemia and chronic lymphocytic leukemia patients demonstrate that PhyloSub can infer both linear (or chain) and branching lineages and its inferences are in good agreement with ground truth, where it is available.
Predicting accurate probabilities with a ranking loss
Jun 18, 2012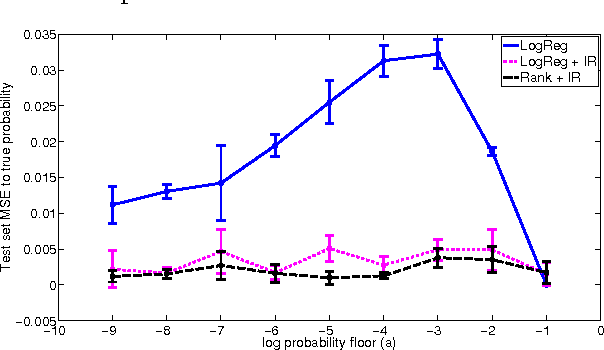
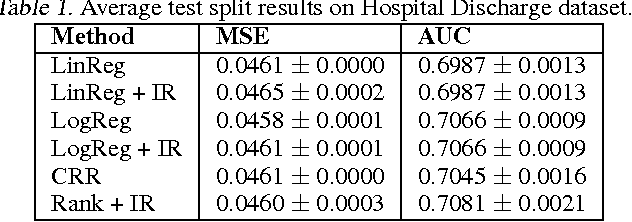
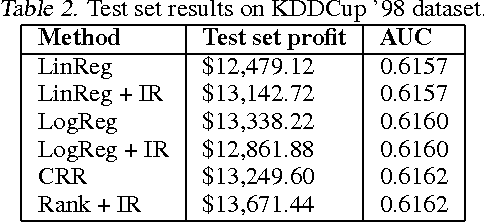
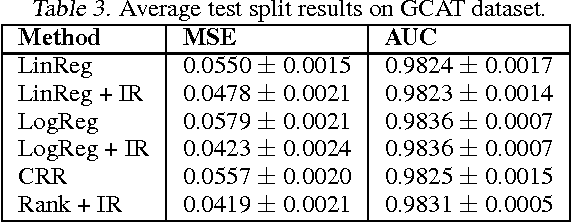
In many real-world applications of machine learning classifiers, it is essential to predict the probability of an example belonging to a particular class. This paper proposes a simple technique for predicting probabilities based on optimizing a ranking loss, followed by isotonic regression. This semi-parametric technique offers both good ranking and regression performance, and models a richer set of probability distributions than statistical workhorses such as logistic regression. We provide experimental results that show the effectiveness of this technique on real-world applications of probability prediction.
Probabilistic Structured Predictors
May 09, 2012We consider MAP estimators for structured prediction with exponential family models. In particular, we concentrate on the case that efficient algorithms for uniform sampling from the output space exist. We show that under this assumption (i) exact computation of the partition function remains a hard problem, and (ii) the partition function and the gradient of the log partition function can be approximated efficiently. Our main result is an approximation scheme for the partition function based on Markov Chain Monte Carlo theory. We also show that the efficient uniform sampling assumption holds in several application settings that are of importance in machine learning.
Learning to Predict Combinatorial Structures
Jun 26, 2010
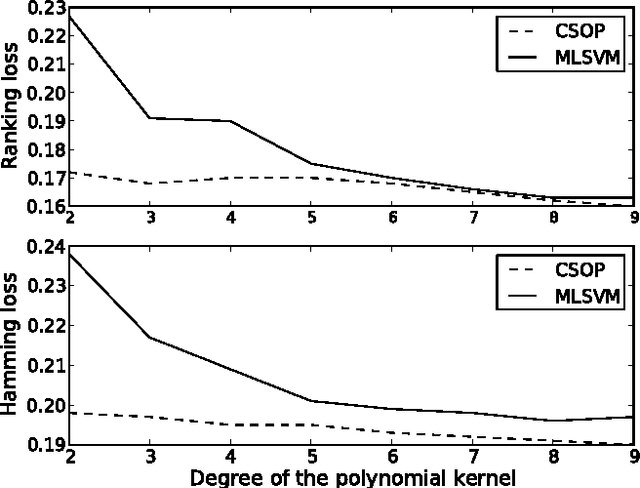
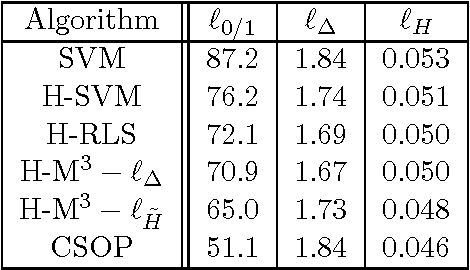
The major challenge in designing a discriminative learning algorithm for predicting structured data is to address the computational issues arising from the exponential size of the output space. Existing algorithms make different assumptions to ensure efficient, polynomial time estimation of model parameters. For several combinatorial structures, including cycles, partially ordered sets, permutations and other graph classes, these assumptions do not hold. In this thesis, we address the problem of designing learning algorithms for predicting combinatorial structures by introducing two new assumptions: (i) The first assumption is that a particular counting problem can be solved efficiently. The consequence is a generalisation of the classical ridge regression for structured prediction. (ii) The second assumption is that a particular sampling problem can be solved efficiently. The consequence is a new technique for designing and analysing probabilistic structured prediction models. These results can be applied to solve several complex learning problems including but not limited to multi-label classification, multi-category hierarchical classification, and label ranking.