 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssociative Memory and Generative Diffusion in the Zero-noise Limit
Jun 05, 2025Connections between generative diffusion and continuous-state associative memory models are studied. Morse-Smale dynamical systems are emphasized as universal approximators of gradient-based associative memory models and diffusion models as white-noise perturbed systems thereof. Universal properties of associative memory that follow from this description are described and used to characterize a generic transition from generation to memory as noise levels diminish. Structural stability inherited by Morse-Smale flows is shown to imply a notion of stability for diffusions at vanishing noise levels. Applied to one- and two-parameter families of gradients, this indicates stability at all but isolated points of associative memory learning landscapes and the learning and generation landscapes of diffusion models with gradient drift in the zero-noise limit, at which small sets of generic bifurcations characterize qualitative transitions between stable systems. Examples illustrating the characterization of these landscapes by sequences of these bifurcations are given, along with structural stability criterion for classic and modern Hopfield networks (equivalently, the attention mechanism).
Learning Optimal Predictive Checklists
Dec 02, 2021


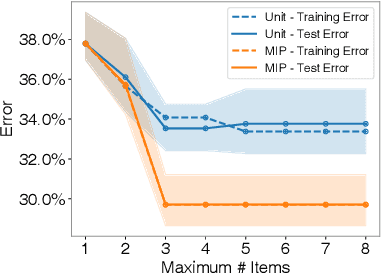
Checklists are simple decision aids that are often used to promote safety and reliability in clinical applications. In this paper, we present a method to learn checklists for clinical decision support. We represent predictive checklists as discrete linear classifiers with binary features and unit weights. We then learn globally optimal predictive checklists from data by solving an integer programming problem. Our method allows users to customize checklists to obey complex constraints, including constraints to enforce group fairness and to binarize real-valued features at training time. In addition, it pairs models with an optimality gap that can inform model development and determine the feasibility of learning sufficiently accurate checklists on a given dataset. We pair our method with specialized techniques that speed up its ability to train a predictive checklist that performs well and has a small optimality gap. We benchmark the performance of our method on seven clinical classification problems, and demonstrate its practical benefits by training a short-form checklist for PTSD screening. Our results show that our method can fit simple predictive checklists that perform well and that can easily be customized to obey a rich class of custom constraints.
Segmenting Hybrid Trajectories using Latent ODEs
May 09, 2021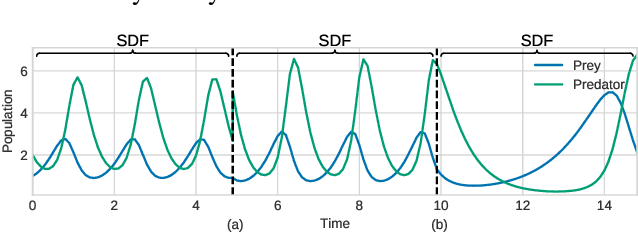
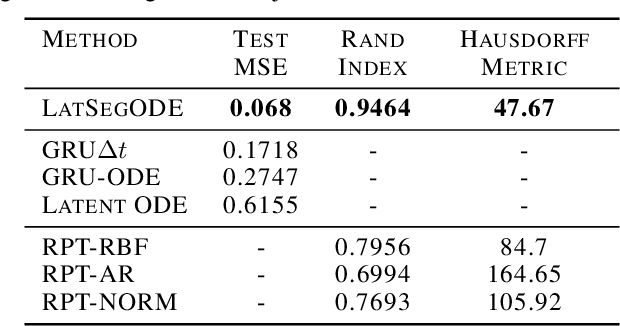
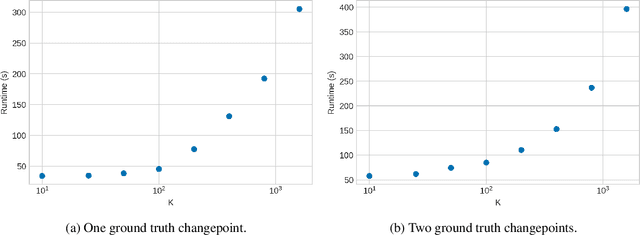
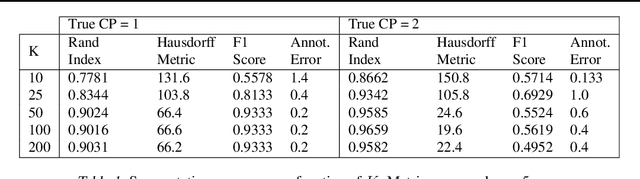
Smooth dynamics interrupted by discontinuities are known as hybrid systems and arise commonly in nature. Latent ODEs allow for powerful representation of irregularly sampled time series but are not designed to capture trajectories arising from hybrid systems. Here, we propose the Latent Segmented ODE (LatSegODE), which uses Latent ODEs to perform reconstruction and changepoint detection within hybrid trajectories featuring jump discontinuities and switching dynamical modes. Where it is possible to train a Latent ODE on the smooth dynamical flows between discontinuities, we apply the pruned exact linear time (PELT) algorithm to detect changepoints where latent dynamics restart, thereby maximizing the joint probability of a piece-wise continuous latent dynamical representation. We propose usage of the marginal likelihood as a score function for PELT, circumventing the need for model complexity-based penalization. The LatSegODE outperforms baselines in reconstructive and segmentation tasks including synthetic data sets of sine waves, Lotka Volterra dynamics, and UCI Character Trajectories.
An Empirical Framework for Domain Generalization in Clinical Settings
Apr 15, 2021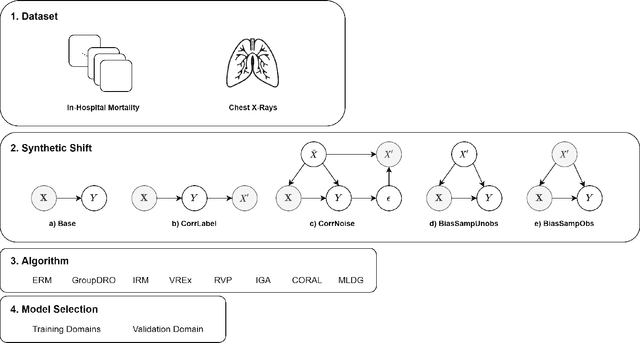
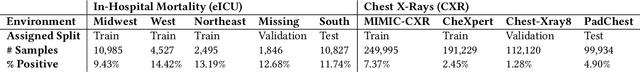
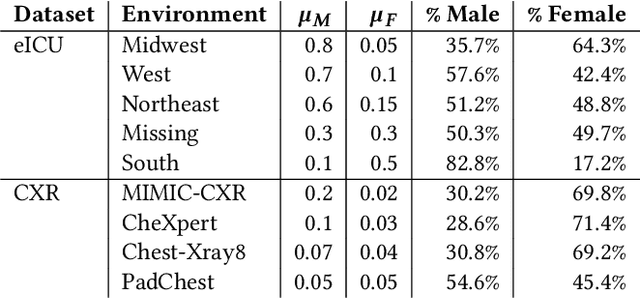
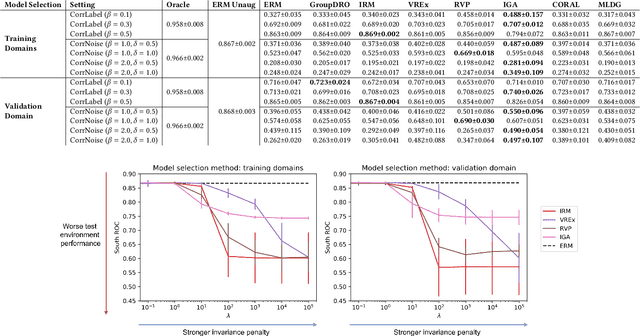
Clinical machine learning models experience significantly degraded performance in datasets not seen during training, e.g., new hospitals or populations. Recent developments in domain generalization offer a promising solution to this problem by creating models that learn invariances across environments. In this work, we benchmark the performance of eight domain generalization methods on multi-site clinical time series and medical imaging data. We introduce a framework to induce synthetic but realistic domain shifts and sampling bias to stress-test these methods over existing non-healthcare benchmarks. We find that current domain generalization methods do not consistently achieve significant gains in out-of-distribution performance over empirical risk minimization on real-world medical imaging data, in line with prior work on general imaging datasets. However, a subset of realistic induced-shift scenarios in clinical time series data do exhibit limited performance gains. We characterize these scenarios in detail, and recommend best practices for domain generalization in the clinical setting.
Memory-Based Graph Networks
Feb 21, 2020



Graph neural networks (GNNs) are a class of deep models that operate on data with arbitrary topology represented as graphs. We introduce an efficient memory layer for GNNs that can jointly learn node representations and coarsen the graph. We also introduce two new networks based on this layer: memory-based GNN (MemGNN) and graph memory network (GMN) that can learn hierarchical graph representations. The experimental results shows that the proposed models achieve state-of-the-art results in eight out of nine graph classification and regression benchmarks. We also show that the learned representations could correspond to chemical features in the molecule data. Code and reference implementations are released at: https://github.com/amirkhas/GraphMemoryNet
Reinterpreting Importance-Weighted Autoencoders
Aug 15, 2017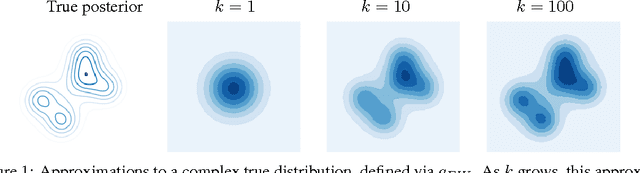
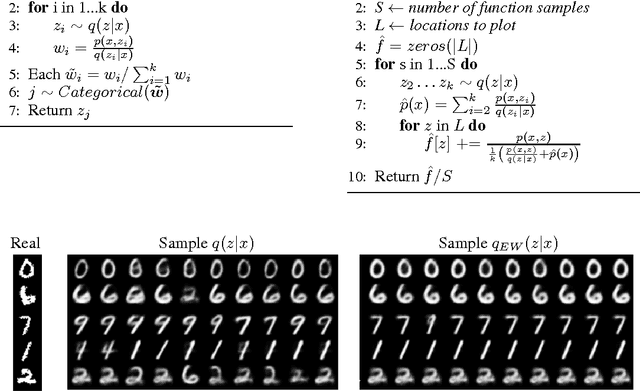
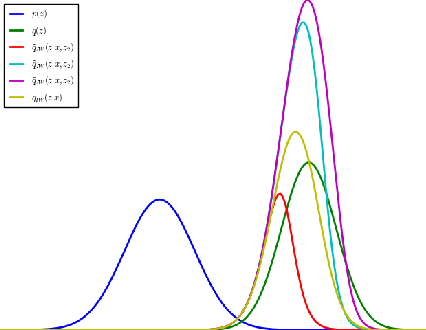
The standard interpretation of importance-weighted autoencoders is that they maximize a tighter lower bound on the marginal likelihood than the standard evidence lower bound. We give an alternate interpretation of this procedure: that it optimizes the standard variational lower bound, but using a more complex distribution. We formally derive this result, present a tighter lower bound, and visualize the implicit importance-weighted distribution.
Reconstructing subclonal composition and evolution from whole genome sequencing of tumors
Jan 06, 2015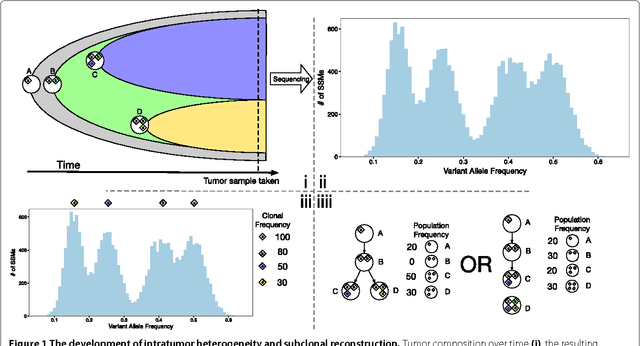
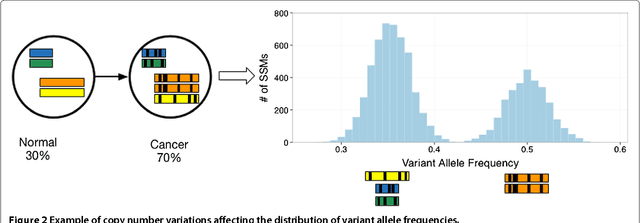
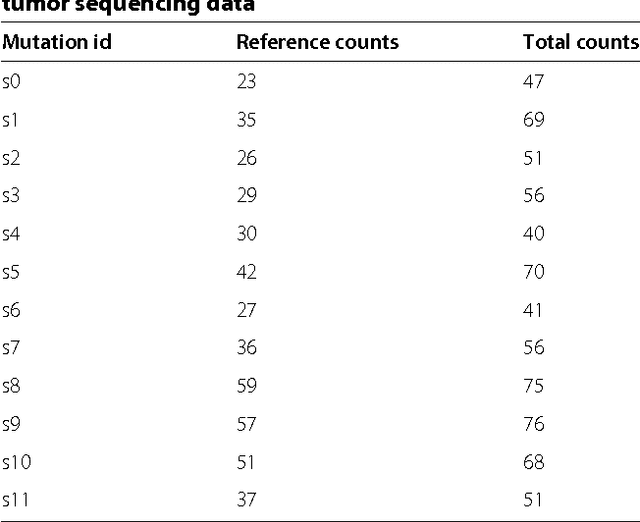
Tumors often contain multiple subpopulations of cancerous cells defined by distinct somatic mutations. We describe a new method, PhyloWGS, that can be applied to WGS data from one or more tumor samples to reconstruct complete genotypes of these subpopulations based on variant allele frequencies (VAFs) of point mutations and population frequencies of structural variations. We introduce a principled phylogenic correction for VAFs in loci affected by copy number alterations and we show that this correction greatly improves subclonal reconstruction compared to existing methods.
Comparing Nonparametric Bayesian Tree Priors for Clonal Reconstruction of Tumors
Aug 11, 2014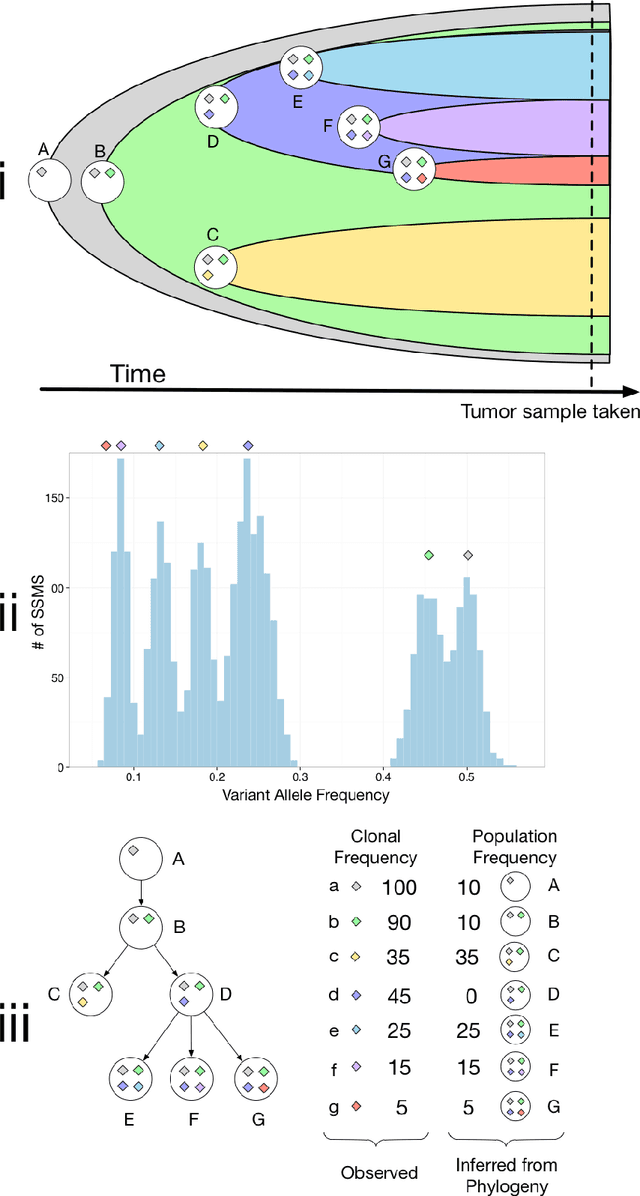
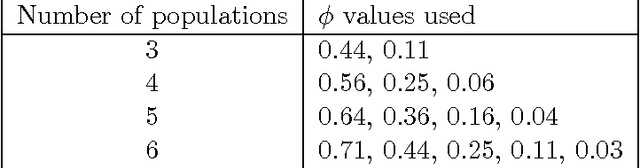
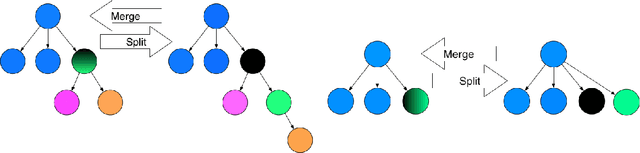
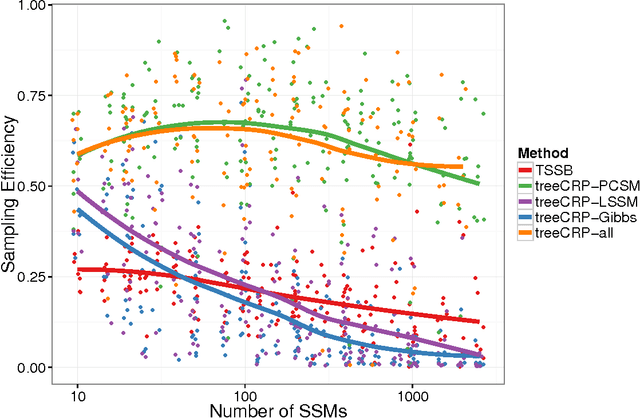
Statistical machine learning methods, especially nonparametric Bayesian methods, have become increasingly popular to infer clonal population structure of tumors. Here we describe the treeCRP, an extension of the Chinese restaurant process (CRP), a popular construction used in nonparametric mixture models, to infer the phylogeny and genotype of major subclonal lineages represented in the population of cancer cells. We also propose new split-merge updates tailored to the subclonal reconstruction problem that improve the mixing time of Markov chains. In comparisons with the tree-structured stick breaking prior used in PhyloSub, we demonstrate superior mixing and running time using the treeCRP with our new split-merge procedures. We also show that given the same number of samples, TSSB and treeCRP have similar ability to recover the subclonal structure of a tumor.
Inferring clonal evolution of tumors from single nucleotide somatic mutations
Nov 02, 2013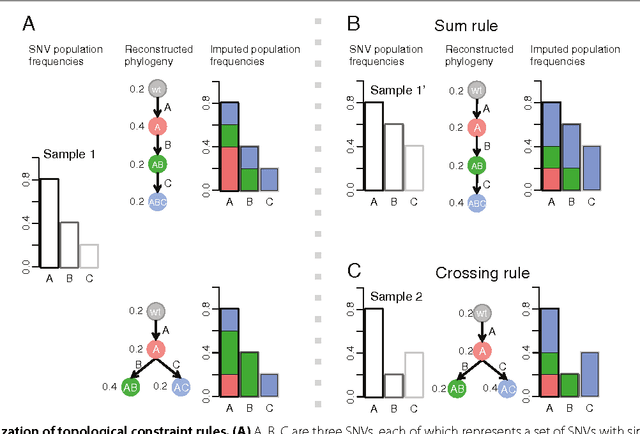
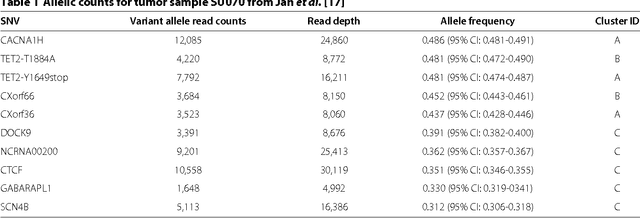
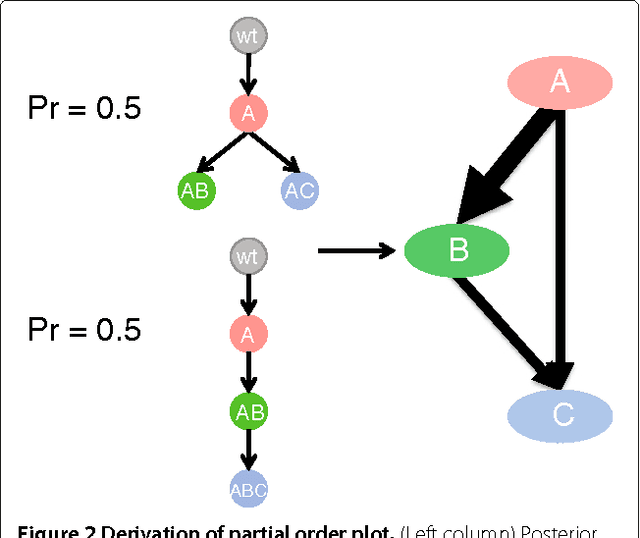
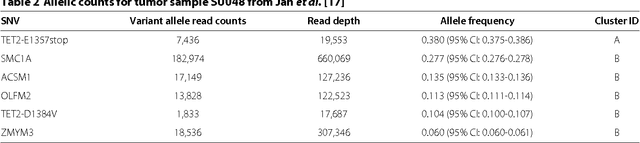
High-throughput sequencing allows the detection and quantification of frequencies of somatic single nucleotide variants (SNV) in heterogeneous tumor cell populations. In some cases, the evolutionary history and population frequency of the subclonal lineages of tumor cells present in the sample can be reconstructed from these SNV frequency measurements. However, automated methods to do this reconstruction are not available and the conditions under which reconstruction is possible have not been described. We describe the conditions under which the evolutionary history can be uniquely reconstructed from SNV frequencies from single or multiple samples from the tumor population and we introduce a new statistical model, PhyloSub, that infers the phylogeny and genotype of the major subclonal lineages represented in the population of cancer cells. It uses a Bayesian nonparametric prior over trees that groups SNVs into major subclonal lineages and automatically estimates the number of lineages and their ancestry. We sample from the joint posterior distribution over trees to identify evolutionary histories and cell population frequencies that have the highest probability of generating the observed SNV frequency data. When multiple phylogenies are consistent with a given set of SNV frequencies, PhyloSub represents the uncertainty in the tumor phylogeny using a partial order plot. Experiments on a simulated dataset and two real datasets comprising tumor samples from acute myeloid leukemia and chronic lymphocytic leukemia patients demonstrate that PhyloSub can infer both linear (or chain) and branching lineages and its inferences are in good agreement with ground truth, where it is available.
Recognition Networks for Approximate Inference in BN20 Networks
Jan 10, 2013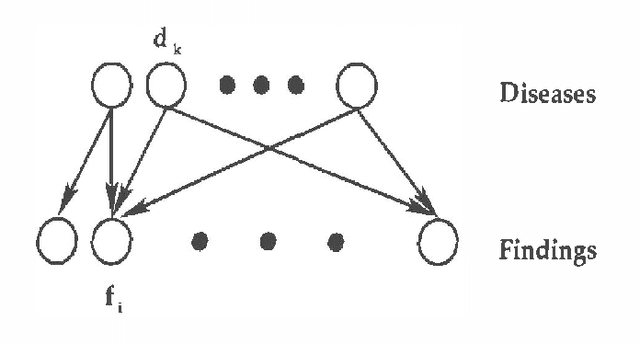
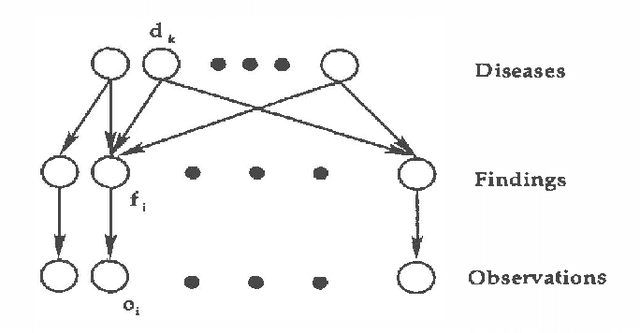
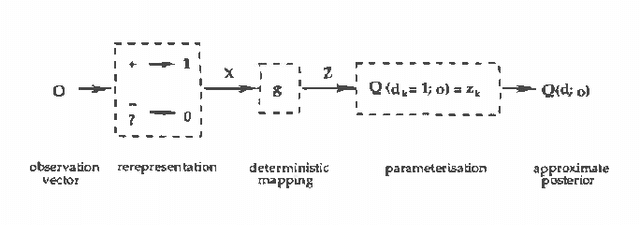
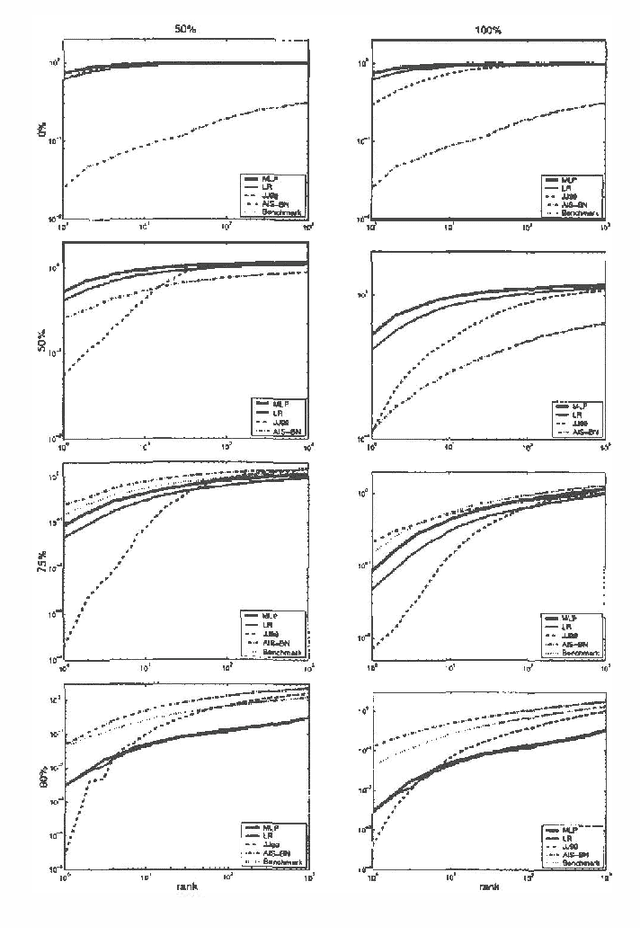
We propose using recognition networks for approximate inference inBayesian networks (BNs). A recognition network is a multilayerperception (MLP) trained to predict posterior marginals given observedevidence in a particular BN. The input to the MLP is a vector of thestates of the evidential nodes. The activity of an output unit isinterpreted as a prediction of the posterior marginal of thecorresponding variable. The MLP is trained using samples generated fromthe corresponding BN.We evaluate a recognition network that was trained to do inference ina large Bayesian network, similar in structure and complexity to theQuick Medical Reference, Decision Theoretic (QMR-DT). Our networkis a binary, two-layer, noisy-OR network containing over 4000 potentially observable nodes and over 600 unobservable, hidden nodes. Inreal medical diagnosis, most observables are unavailable, and there isa complex and unknown bias that selects which ones are provided. Weincorporate a very basic type of selection bias in our network: a knownpreference that available observables are positive rather than negative.Even this simple bias has a significant effect on the posterior. We compare the performance of our recognition network tostate-of-the-art approximate inference algorithms on a large set oftest cases. In order to evaluate the effect of our simplistic modelof the selection bias, we evaluate algorithms using a variety ofincorrectly modeled observation biases. Recognition networks performwell using both correct and incorrect observation biases.