 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInferring clonal evolution of tumors from single nucleotide somatic mutations
Nov 02, 2013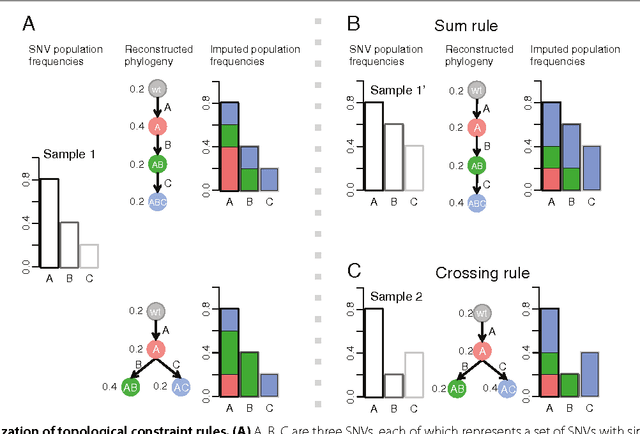
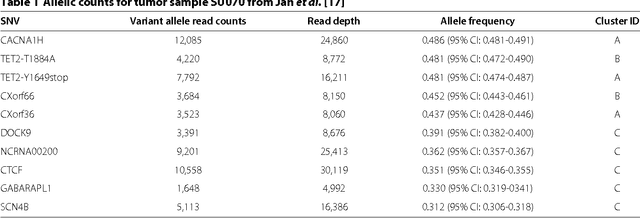
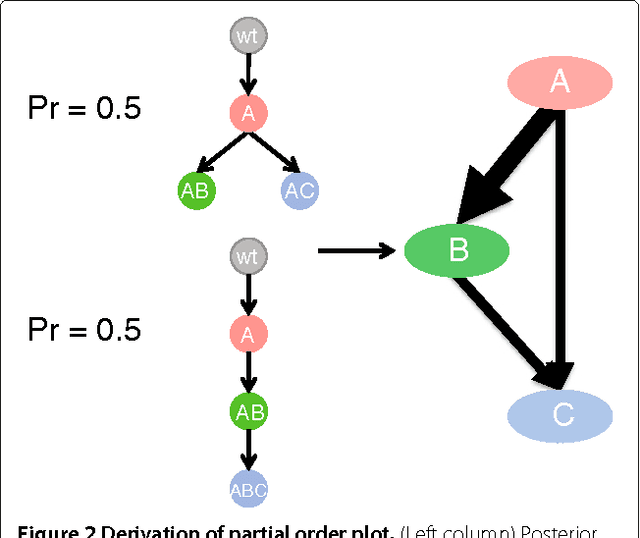
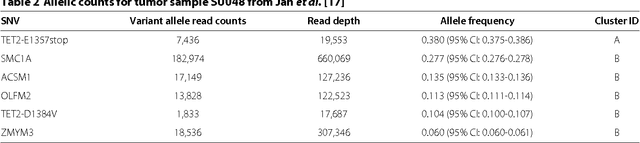
High-throughput sequencing allows the detection and quantification of frequencies of somatic single nucleotide variants (SNV) in heterogeneous tumor cell populations. In some cases, the evolutionary history and population frequency of the subclonal lineages of tumor cells present in the sample can be reconstructed from these SNV frequency measurements. However, automated methods to do this reconstruction are not available and the conditions under which reconstruction is possible have not been described. We describe the conditions under which the evolutionary history can be uniquely reconstructed from SNV frequencies from single or multiple samples from the tumor population and we introduce a new statistical model, PhyloSub, that infers the phylogeny and genotype of the major subclonal lineages represented in the population of cancer cells. It uses a Bayesian nonparametric prior over trees that groups SNVs into major subclonal lineages and automatically estimates the number of lineages and their ancestry. We sample from the joint posterior distribution over trees to identify evolutionary histories and cell population frequencies that have the highest probability of generating the observed SNV frequency data. When multiple phylogenies are consistent with a given set of SNV frequencies, PhyloSub represents the uncertainty in the tumor phylogeny using a partial order plot. Experiments on a simulated dataset and two real datasets comprising tumor samples from acute myeloid leukemia and chronic lymphocytic leukemia patients demonstrate that PhyloSub can infer both linear (or chain) and branching lineages and its inferences are in good agreement with ground truth, where it is available.