 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCensoring-Aware Tree-Based Reinforcement Learning for Estimating Dynamic Treatment Regimes with Censored Outcomes
Mar 09, 2025Dynamic Treatment Regimes (DTRs) provide a systematic approach for making sequential treatment decisions that adapt to individual patient characteristics, particularly in clinical contexts where survival outcomes are of interest. Censoring-Aware Tree-Based Reinforcement Learning (CA-TRL) is a novel framework to address the complexities associated with censored data when estimating optimal DTRs. We explore ways to learn effective DTRs, from observational data. By enhancing traditional tree-based reinforcement learning methods with augmented inverse probability weighting (AIPW) and censoring-aware modifications, CA-TRL delivers robust and interpretable treatment strategies. We demonstrate its effectiveness through extensive simulations and real-world applications using the SANAD epilepsy dataset, where it outperformed the recently proposed ASCL method in key metrics such as restricted mean survival time (RMST) and decision-making accuracy. This work represents a step forward in advancing personalized and data-driven treatment strategies across diverse healthcare settings.
Practical Evaluation of Copula-based Survival Metrics: Beyond the Independent Censoring Assumption
Feb 26, 2025



Conventional survival metrics, such as Harrell's concordance index and the Brier Score, rely on the independent censoring assumption for valid inference in the presence of right-censored data. However, when instances are censored for reasons related to the event of interest, this assumption no longer holds, as this kind of dependent censoring biases the marginal survival estimates of popular nonparametric estimators. In this paper, we propose three copula-based metrics to evaluate survival models in the presence of dependent censoring, and design a framework to create realistic, semi-synthetic datasets with dependent censoring to facilitate the evaluation of the metrics. Our empirical analyses in synthetic and semi-synthetic datasets show that our metrics can give error estimates that are closer to the true error, mainly in terms of predictive accuracy.
Deep Learning for Disease Outbreak Prediction: A Robust Early Warning Signal for Transcritical Bifurcations
Jan 14, 2025



Early Warning Signals (EWSs) are vital for implementing preventive measures before a disease turns into a pandemic. While new diseases exhibit unique behaviors, they often share fundamental characteristics from a dynamical systems perspective. Moreover, measurements during disease outbreaks are often corrupted by different noise sources, posing challenges for Time Series Classification (TSC) tasks. In this study, we address the problem of having a robust EWS for disease outbreak prediction using a best-performing deep learning model in the domain of TSC. We employed two simulated datasets to train the model: one representing generated dynamical systems with randomly selected polynomial terms to model new disease behaviors, and another simulating noise-induced disease dynamics to account for noisy measurements. The model's performance was analyzed using both simulated data from different disease models and real-world data, including influenza and COVID-19. Results demonstrate that the proposed model outperforms previous models, effectively providing EWSs of impending outbreaks across various scenarios. This study bridges advancements in deep learning with the ability to provide robust early warning signals in noisy environments, making it highly applicable to real-world crises involving emerging disease outbreaks.
MassSpecGym: A benchmark for the discovery and identification of molecules
Oct 30, 2024The discovery and identification of molecules in biological and environmental samples is crucial for advancing biomedical and chemical sciences. Tandem mass spectrometry (MS/MS) is the leading technique for high-throughput elucidation of molecular structures. However, decoding a molecular structure from its mass spectrum is exceptionally challenging, even when performed by human experts. As a result, the vast majority of acquired MS/MS spectra remain uninterpreted, thereby limiting our understanding of the underlying (bio)chemical processes. Despite decades of progress in machine learning applications for predicting molecular structures from MS/MS spectra, the development of new methods is severely hindered by the lack of standard datasets and evaluation protocols. To address this problem, we propose MassSpecGym -- the first comprehensive benchmark for the discovery and identification of molecules from MS/MS data. Our benchmark comprises the largest publicly available collection of high-quality labeled MS/MS spectra and defines three MS/MS annotation challenges: \textit{de novo} molecular structure generation, molecule retrieval, and spectrum simulation. It includes new evaluation metrics and a generalization-demanding data split, therefore standardizing the MS/MS annotation tasks and rendering the problem accessible to the broad machine learning community. MassSpecGym is publicly available at \url{https://github.com/pluskal-lab/MassSpecGym}.
Toward Conditional Distribution Calibration in Survival Prediction
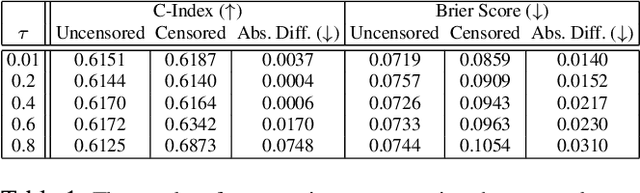
Oct 27, 2024Survival prediction often involves estimating the time-to-event distribution from censored datasets. Previous approaches have focused on enhancing discrimination and marginal calibration. In this paper, we highlight the significance of conditional calibration for real-world applications -- especially its role in individual decision-making. We propose a method based on conformal prediction that uses the model's predicted individual survival probability at that instance's observed time. This method effectively improves the model's marginal and conditional calibration, without compromising discrimination. We provide asymptotic theoretical guarantees for both marginal and conditional calibration and test it extensively across 15 diverse real-world datasets, demonstrating the method's practical effectiveness and versatility in various settings.
MENSA: A Multi-Event Network for Survival Analysis under Informative Censoring
Sep 10, 2024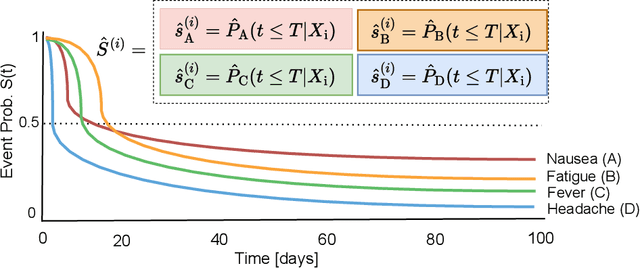

Given an instance, a multi-event survival model predicts the time until that instance experiences each of several different events. These events are not mutually exclusive and there are often statistical dependencies between them. There are relatively few multi-event survival results, most focusing on producing a simple risk score, rather than the time-to-event itself. To overcome these issues, we introduce MENSA, a novel, deep learning approach for multi-event survival analysis that can jointly learn representations of the input covariates and the dependence structure between events. As a practical motivation for multi-event survival analysis, we consider the problem of predicting the time until a patient with amyotrophic lateral sclerosis (ALS) loses various physical functions, i.e., the ability to speak, swallow, write, or walk. When estimating when a patient is no longer able to swallow, our approach achieves an L1-Margin loss of 278.8 days, compared to 355.2 days when modeling each event separately. In addition, we also evaluate our approach in single-event and competing risk scenarios by modeling the censoring and event distributions as equal contributing factors in the optimization process, and show that our approach performs well across multiple benchmark datasets. The source code is available at: https://github.com/thecml/mensa
Conformalized Survival Distributions: A Generic Post-Process to Increase Calibration
May 12, 2024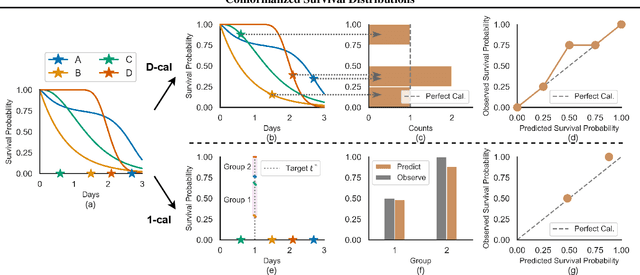
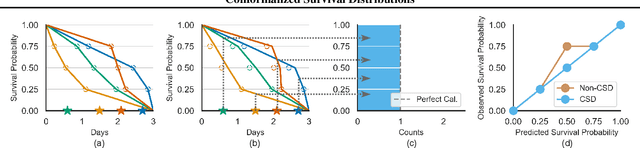

Discrimination and calibration represent two important properties of survival analysis, with the former assessing the model's ability to accurately rank subjects and the latter evaluating the alignment of predicted outcomes with actual events. With their distinct nature, it is hard for survival models to simultaneously optimize both of them especially as many previous results found improving calibration tends to diminish discrimination performance. This paper introduces a novel approach utilizing conformal regression that can improve a model's calibration without degrading discrimination. We provide theoretical guarantees for the above claim, and rigorously validate the efficiency of our approach across 11 real-world datasets, showcasing its practical applicability and robustness in diverse scenarios.
Early detection of disease outbreaks and non-outbreaks using incidence data
Apr 13, 2024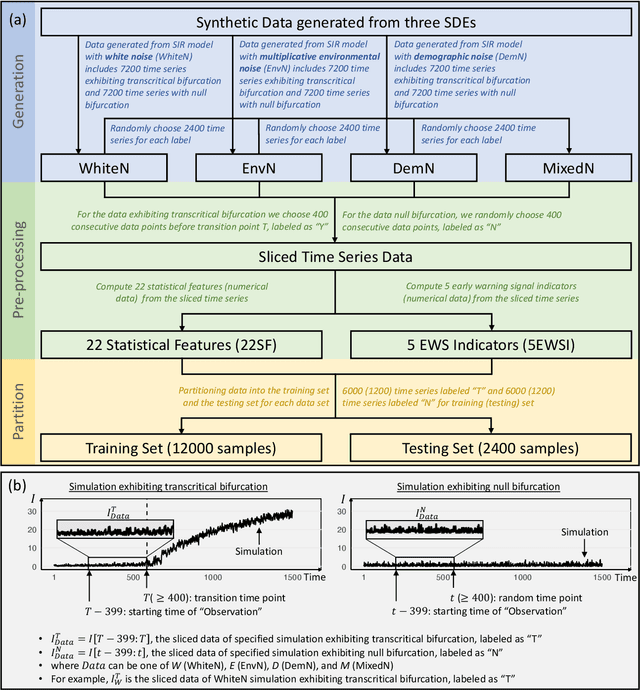
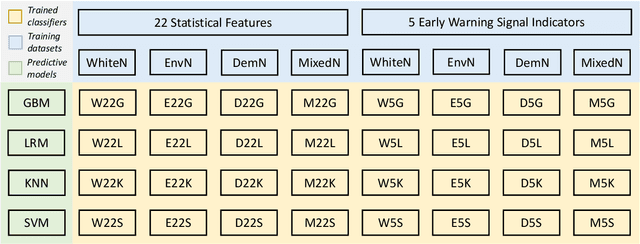
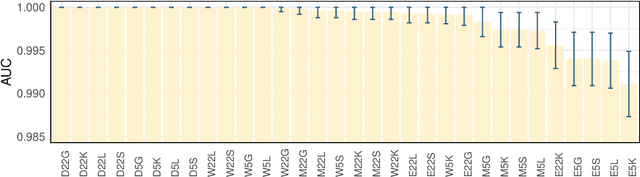
Forecasting the occurrence and absence of novel disease outbreaks is essential for disease management. Here, we develop a general model, with no real-world training data, that accurately forecasts outbreaks and non-outbreaks. We propose a novel framework, using a feature-based time series classification method to forecast outbreaks and non-outbreaks. We tested our methods on synthetic data from a Susceptible-Infected-Recovered model for slowly changing, noisy disease dynamics. Outbreak sequences give a transcritical bifurcation within a specified future time window, whereas non-outbreak (null bifurcation) sequences do not. We identified incipient differences in time series of infectives leading to future outbreaks and non-outbreaks. These differences are reflected in 22 statistical features and 5 early warning signal indicators. Classifier performance, given by the area under the receiver-operating curve, ranged from 0.99 for large expanding windows of training data to 0.7 for small rolling windows. Real-world performances of classifiers were tested on two empirical datasets, COVID-19 data from Singapore and SARS data from Hong Kong, with two classifiers exhibiting high accuracy. In summary, we showed that there are statistical features that distinguish outbreak and non-outbreak sequences long before outbreaks occur. We could detect these differences in synthetic and real-world data sets, well before potential outbreaks occur.
An early warning indicator trained on stochastic disease-spreading models with different noises
Mar 24, 2024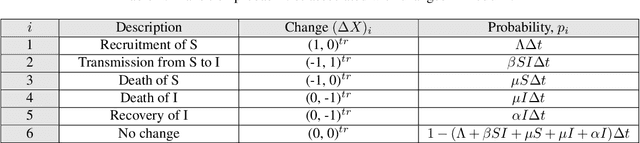
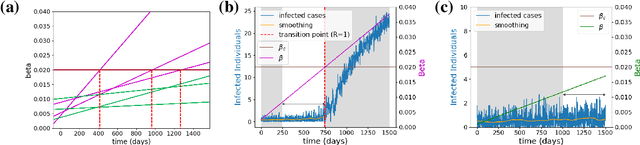
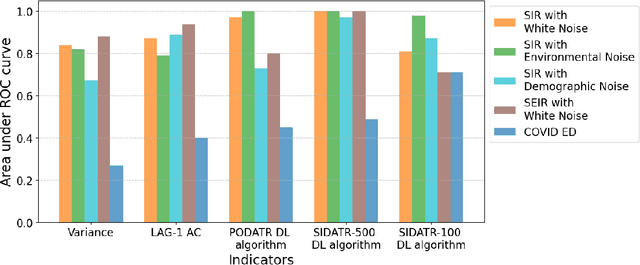
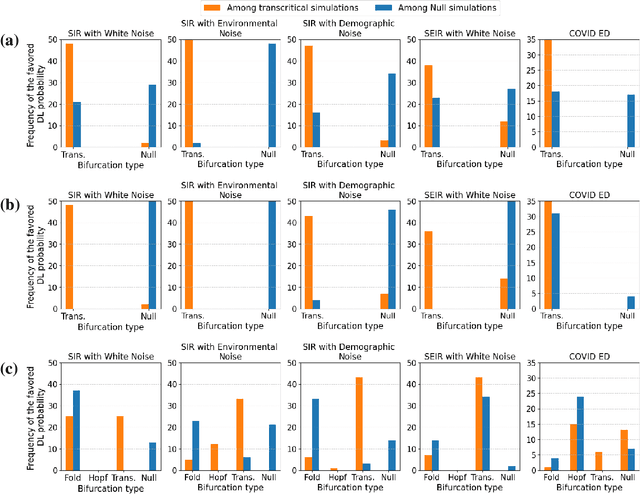
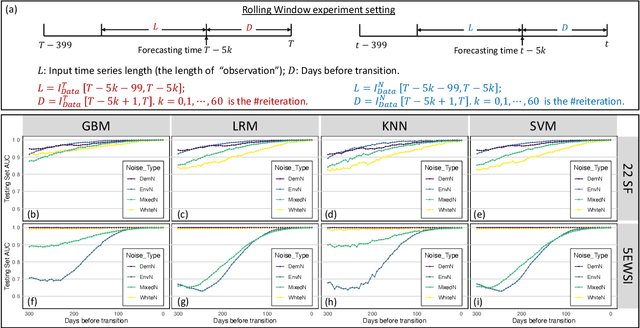
The timely detection of disease outbreaks through reliable early warning signals (EWSs) is indispensable for effective public health mitigation strategies. Nevertheless, the intricate dynamics of real-world disease spread, often influenced by diverse sources of noise and limited data in the early stages of outbreaks, pose a significant challenge in developing reliable EWSs, as the performance of existing indicators varies with extrinsic and intrinsic noises. Here, we address the challenge of modeling disease when the measurements are corrupted by additive white noise, multiplicative environmental noise, and demographic noise into a standard epidemic mathematical model. To navigate the complexities introduced by these noise sources, we employ a deep learning algorithm that provides EWS in infectious disease outbreak by training on noise-induced disease-spreading models. The indicator's effectiveness is demonstrated through its application to real-world COVID-19 cases in Edmonton and simulated time series derived from diverse disease spread models affected by noise. Notably, the indicator captures an impending transition in a time series of disease outbreaks and outperforms existing indicators. This study contributes to advancing early warning capabilities by addressing the intricate dynamics inherent in real-world disease spread, presenting a promising avenue for enhancing public health preparedness and response efforts.
Copula-Based Deep Survival Models for Dependent Censoring
Jun 20, 2023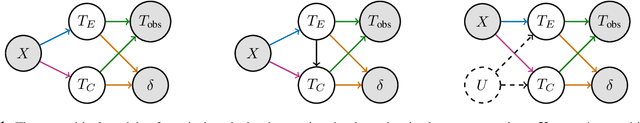
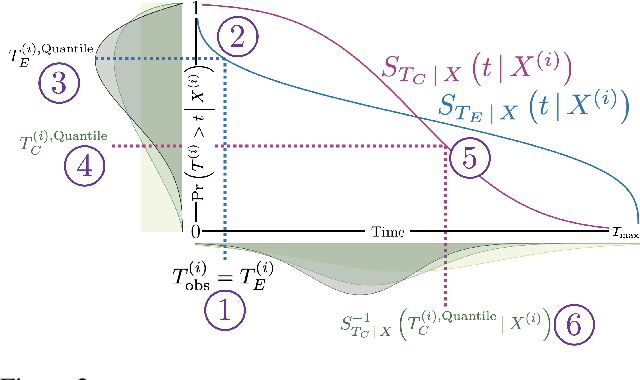
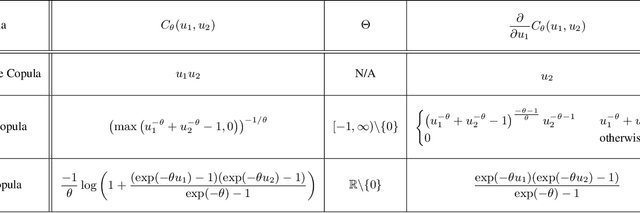
A survival dataset describes a set of instances (e.g. patients) and provides, for each, either the time until an event (e.g. death), or the censoring time (e.g. when lost to follow-up - which is a lower bound on the time until the event). We consider the challenge of survival prediction: learning, from such data, a predictive model that can produce an individual survival distribution for a novel instance. Many contemporary methods of survival prediction implicitly assume that the event and censoring distributions are independent conditional on the instance's covariates - a strong assumption that is difficult to verify (as we observe only one outcome for each instance) and which can induce significant bias when it does not hold. This paper presents a parametric model of survival that extends modern non-linear survival analysis by relaxing the assumption of conditional independence. On synthetic and semi-synthetic data, our approach significantly improves estimates of survival distributions compared to the standard that assumes conditional independence in the data.