 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMeasuring Perceived Trust in XAI-Assisted Decision-Making by Eliciting a Mental Model
Jul 15, 2023



This empirical study proposes a novel methodology to measure users' perceived trust in an Explainable Artificial Intelligence (XAI) model. To do so, users' mental models are elicited using Fuzzy Cognitive Maps (FCMs). First, we exploit an interpretable Machine Learning (ML) model to classify suspected COVID-19 patients into positive or negative cases. Then, Medical Experts' (MEs) conduct a diagnostic decision-making task based on their knowledge and then prediction and interpretations provided by the XAI model. In order to evaluate the impact of interpretations on perceived trust, explanation satisfaction attributes are rated by MEs through a survey. Then, they are considered as FCM's concepts to determine their influences on each other and, ultimately, on the perceived trust. Moreover, to consider MEs' mental subjectivity, fuzzy linguistic variables are used to determine the strength of influences. After reaching the steady state of FCMs, a quantified value is obtained to measure the perceived trust of each ME. The results show that the quantified values can determine whether MEs trust or distrust the XAI model. We analyze this behavior by comparing the quantified values with MEs' performance in completing diagnostic tasks.
Assisting clinical practice with fuzzy probabilistic decision trees
Apr 26, 2023The need for fully human-understandable models is increasingly being recognised as a central theme in AI research. The acceptance of AI models to assist in decision making in sensitive domains will grow when these models are interpretable, and this trend towards interpretable models will be amplified by upcoming regulations. One of the killer applications of interpretable AI is medical practice, which can benefit from accurate decision support methodologies that inherently generate trust. In this work, we propose FPT, (MedFP), a novel method that combines probabilistic trees and fuzzy logic to assist clinical practice. This approach is fully interpretable as it allows clinicians to generate, control and verify the entire diagnosis procedure; one of the methodology's strength is the capability to decrease the frequency of misdiagnoses by providing an estimate of uncertainties and counterfactuals. Our approach is applied as a proof-of-concept to two real medical scenarios: classifying malignant thyroid nodules and predicting the risk of progression in chronic kidney disease patients. Our results show that probabilistic fuzzy decision trees can provide interpretable support to clinicians, furthermore, introducing fuzzy variables into the probabilistic model brings significant nuances that are lost when using the crisp thresholds set by traditional probabilistic decision trees. We show that FPT and its predictions can assist clinical practice in an intuitive manner, with the use of a user-friendly interface specifically designed for this purpose. Moreover, we discuss the interpretability of the FPT model.
Salp Swarm Optimization: a Critical Review
Jun 03, 2021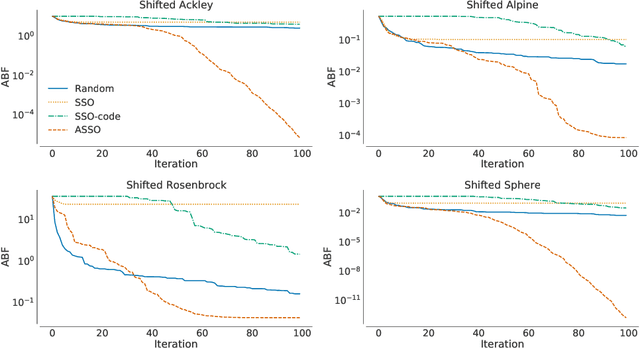
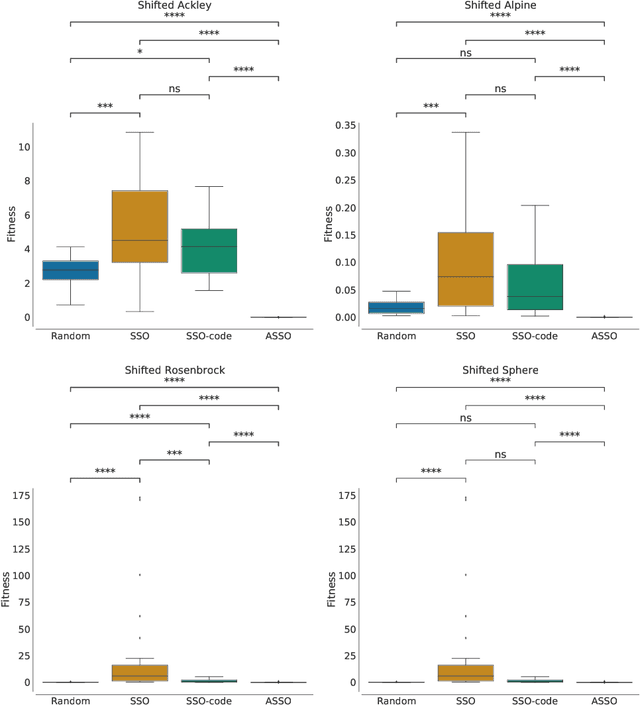
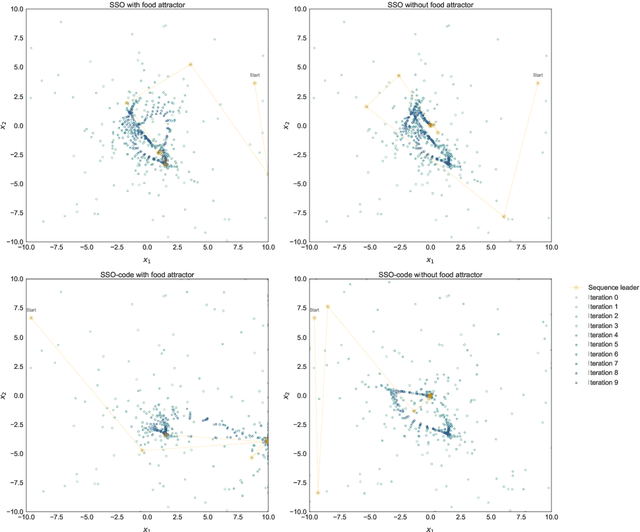
In the crowded environment of bio-inspired population-based meta-heuristics, the Salp Swarm Optimization (SSO) algorithm recently appeared and immediately gained a lot of momentum. Inspired by the peculiar spatial arrangement of salp colonies, which are displaced in long chains following a leader, this algorithm seems to provide interesting optimization performances. However, the original work was characterized by some conceptual and mathematical flaws, which influenced all ensuing papers on the subject. In this manuscript, we perform a critical review of SSO, highlighting all the issues present in the literature and their negative effects on the optimization process carried out by the algorithm. We also propose a mathematically correct version of SSO, named Amended Salp Swarm Optimizer (ASSO) that fixes all the discussed problems. Finally, we benchmark the performance of ASSO on a set of tailored experiments, showing it achieves better results than the original SSO.
USE-Net: incorporating Squeeze-and-Excitation blocks into U-Net for prostate zonal segmentation of multi-institutional MRI datasets
Apr 17, 2019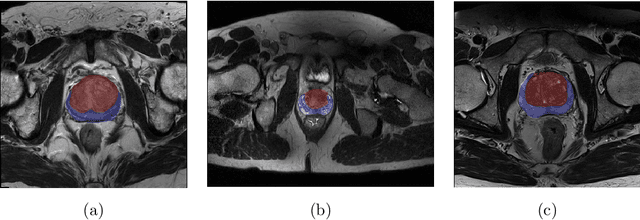
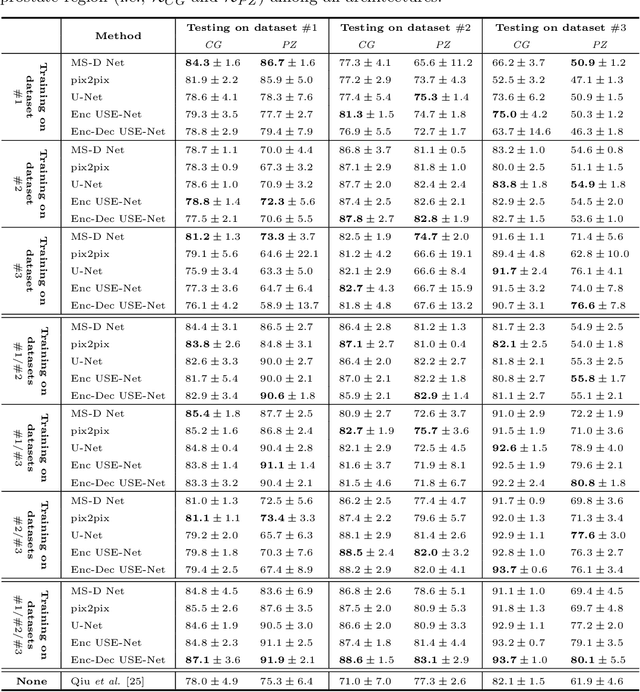
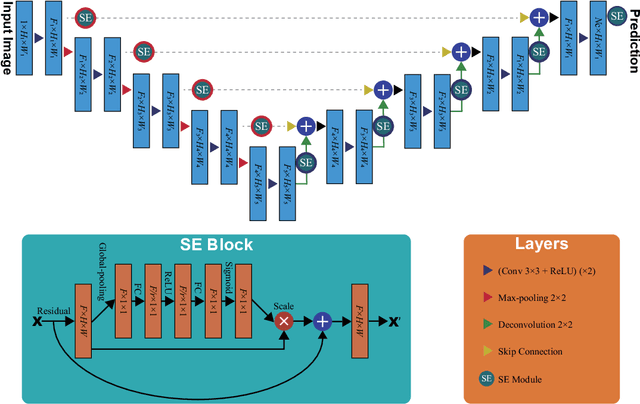
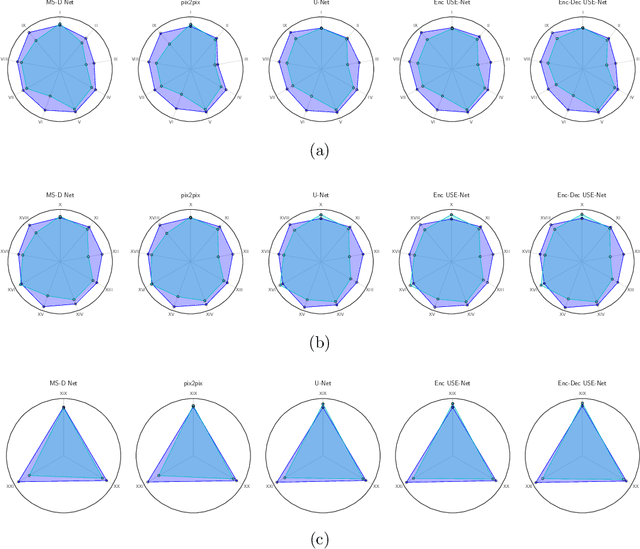
Prostate cancer is the most common malignant tumors in men but prostate Magnetic Resonance Imaging (MRI) analysis remains challenging. Besides whole prostate gland segmentation, the capability to differentiate between the blurry boundary of the Central Gland (CG) and Peripheral Zone (PZ) can lead to differential diagnosis, since tumor's frequency and severity differ in these regions. To tackle the prostate zonal segmentation task, we propose a novel Convolutional Neural Network (CNN), called USE-Net, which incorporates Squeeze-and-Excitation (SE) blocks into U-Net. Especially, the SE blocks are added after every Encoder (Enc USE-Net) or Encoder-Decoder block (Enc-Dec USE-Net). This study evaluates the generalization ability of CNN-based architectures on three T2-weighted MRI datasets, each one consisting of a different number of patients and heterogeneous image characteristics, collected by different institutions. The following mixed scheme is used for training/testing: (i) training on either each individual dataset or multiple prostate MRI datasets and (ii) testing on all three datasets with all possible training/testing combinations. USE-Net is compared against three state-of-the-art CNN-based architectures (i.e., U-Net, pix2pix, and Mixed-Scale Dense Network), along with a semi-automatic continuous max-flow model. The results show that training on the union of the datasets generally outperforms training on each dataset separately, allowing for both intra-/cross-dataset generalization. Enc USE-Net shows good overall generalization under any training condition, while Enc-Dec USE-Net remarkably outperforms the other methods when trained on all datasets. These findings reveal that the SE blocks' adaptive feature recalibration provides excellent cross-dataset generalization when testing is performed on samples of the datasets used during training.
CNN-based Prostate Zonal Segmentation on T2-weighted MR Images: A Cross-dataset Study
Mar 29, 2019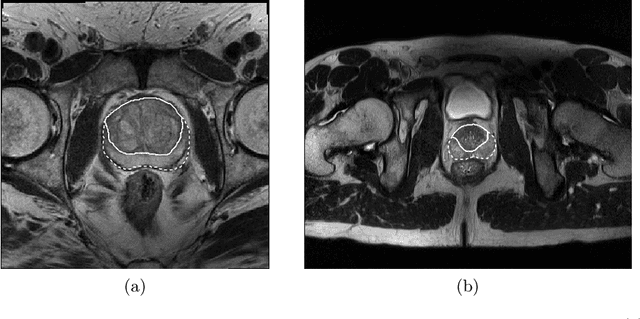
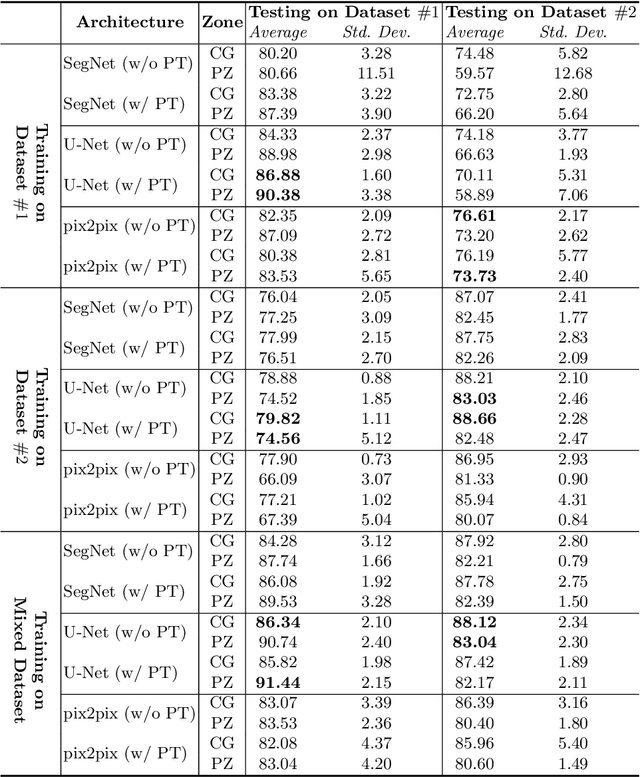
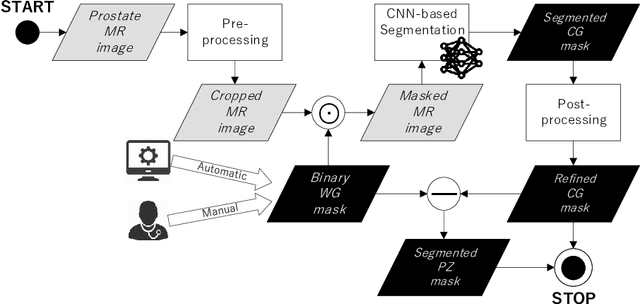
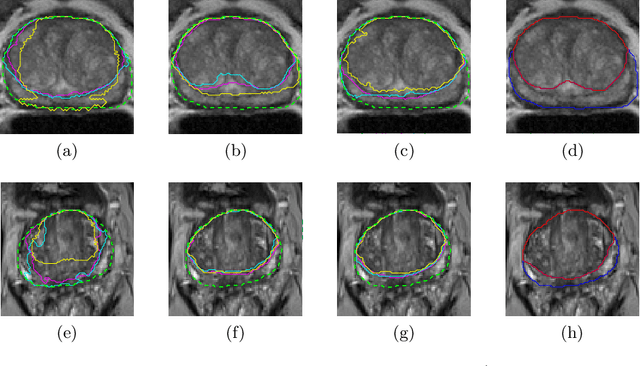
Prostate cancer is the most common cancer among US men. However, prostate imaging is still challenging despite the advances in multi-parametric Magnetic Resonance Imaging (MRI), which provides both morphologic and functional information pertaining to the pathological regions. Along with whole prostate gland segmentation, distinguishing between the Central Gland (CG) and Peripheral Zone (PZ) can guide towards differential diagnosis, since the frequency and severity of tumors differ in these regions; however, their boundary is often weak and fuzzy. This work presents a preliminary study on Deep Learning to automatically delineate the CG and PZ, aiming at evaluating the generalization ability of Convolutional Neural Networks (CNNs) on two multi-centric MRI prostate datasets. Especially, we compared three CNN-based architectures: SegNet, U-Net, and pix2pix. In such a context, the segmentation performances achieved with/without pre-training were compared in 4-fold cross-validation. In general, U-Net outperforms the other methods, especially when training and testing are performed on multiple datasets.
Efficient computational strategies to learn the structure of probabilistic graphical models of cumulative phenomena
Oct 23, 2018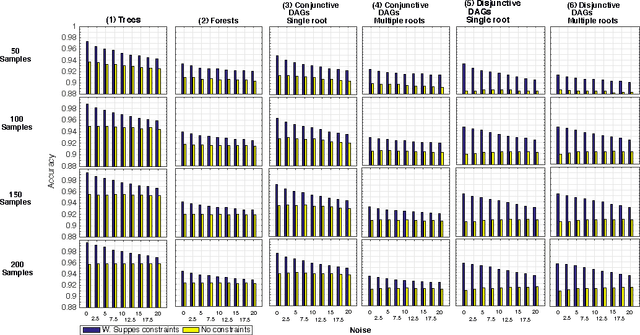
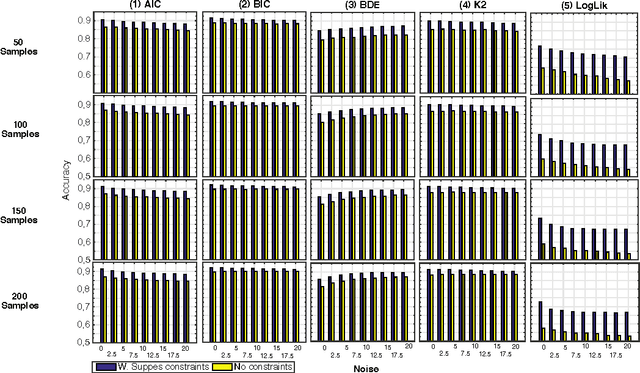
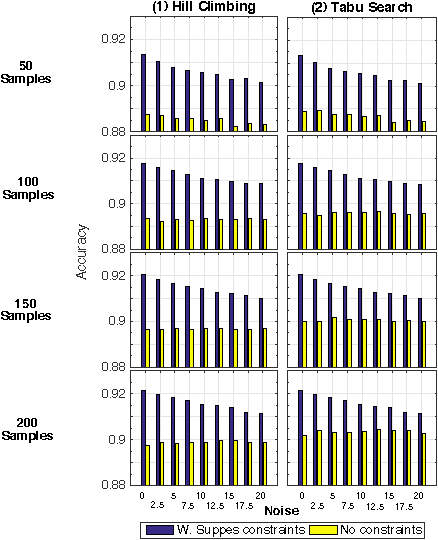
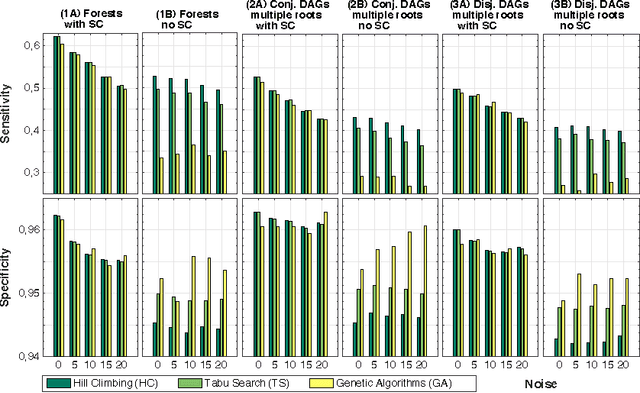
Structural learning of Bayesian Networks (BNs) is a NP-hard problem, which is further complicated by many theoretical issues, such as the I-equivalence among different structures. In this work, we focus on a specific subclass of BNs, named Suppes-Bayes Causal Networks (SBCNs), which include specific structural constraints based on Suppes' probabilistic causation to efficiently model cumulative phenomena. Here we compare the performance, via extensive simulations, of various state-of-the-art search strategies, such as local search techniques and Genetic Algorithms, as well as of distinct regularization methods. The assessment is performed on a large number of simulated datasets from topologies with distinct levels of complexity, various sample size and different rates of errors in the data. Among the main results, we show that the introduction of Suppes' constraints dramatically improve the inference accuracy, by reducing the solution space and providing a temporal ordering on the variables. We also report on trade-offs among different search techniques that can be efficiently employed in distinct experimental settings. This manuscript is an extended version of the paper "Structural Learning of Probabilistic Graphical Models of Cumulative Phenomena" presented at the 2018 International Conference on Computational Science.
Multi-objective optimization to explicitly account for model complexity when learning Bayesian Networks
Aug 03, 2018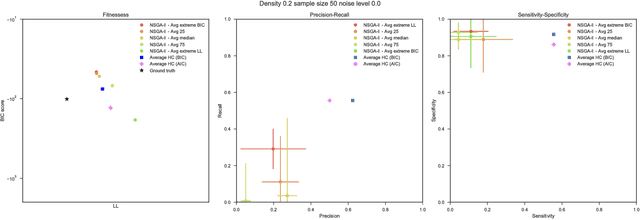
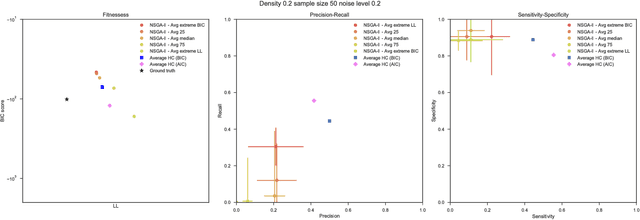
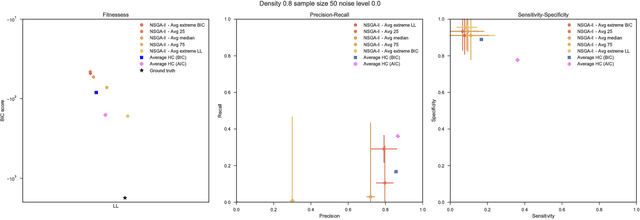
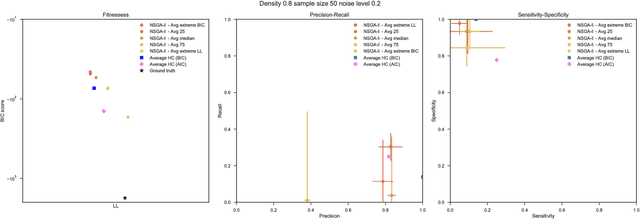
Bayesian Networks have been widely used in the last decades in many fields, to describe statistical dependencies among random variables. In general, learning the structure of such models is a problem with considerable theoretical interest that still poses many challenges. On the one hand, this is a well-known NP-complete problem, which is practically hardened by the huge search space of possible solutions. On the other hand, the phenomenon of I-equivalence, i.e., different graphical structures underpinning the same set of statistical dependencies, may lead to multimodal fitness landscapes further hindering maximum likelihood approaches to solve the task. Despite all these difficulties, greedy search methods based on a likelihood score coupled with a regularization term to account for model complexity, have been shown to be surprisingly effective in practice. In this paper, we consider the formulation of the task of learning the structure of Bayesian Networks as an optimization problem based on a likelihood score. Nevertheless, our approach do not adjust this score by means of any of the complexity terms proposed in the literature; instead, it accounts directly for the complexity of the discovered solutions by exploiting a multi-objective optimization procedure. To this extent, we adopt NSGA-II and define the first objective function to be the likelihood of a solution and the second to be the number of selected arcs. We thoroughly analyze the behavior of our method on a wide set of simulated data, and we discuss the performance considering the goodness of the inferred solutions both in terms of their objective functions and with respect to the retrieved structure. Our results show that NSGA-II can converge to solutions characterized by better likelihood and less arcs than classic approaches, although paradoxically frequently characterized by a lower similarity to the target network.
Parallel Implementation of Efficient Search Schemes for the Inference of Cancer Progression Models
Mar 08, 2017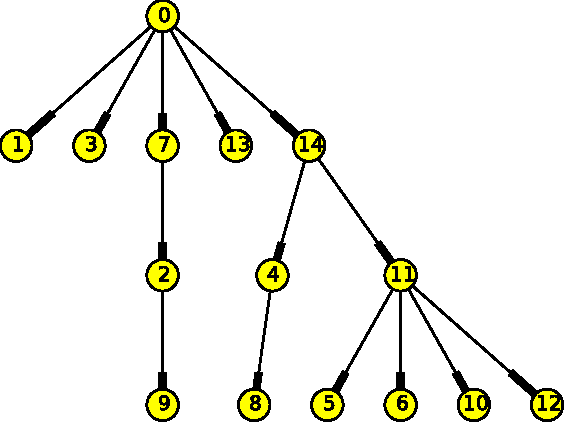
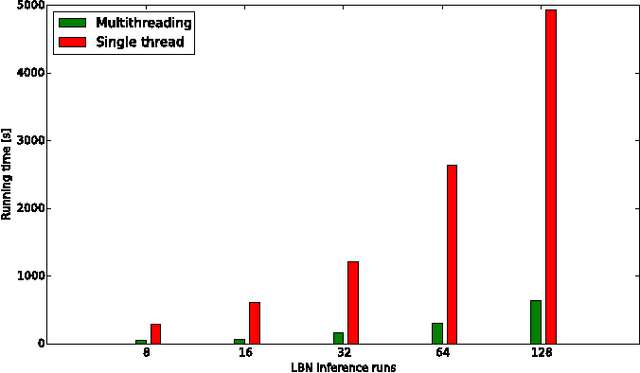
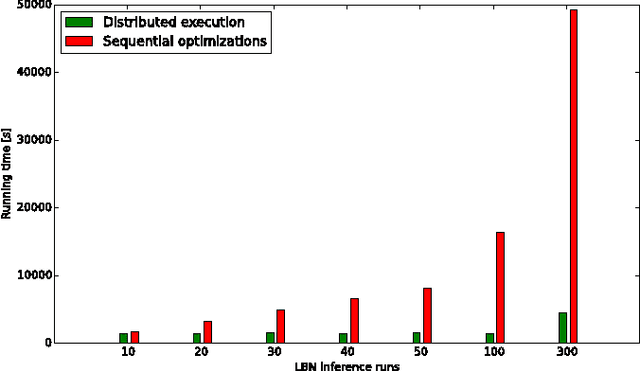
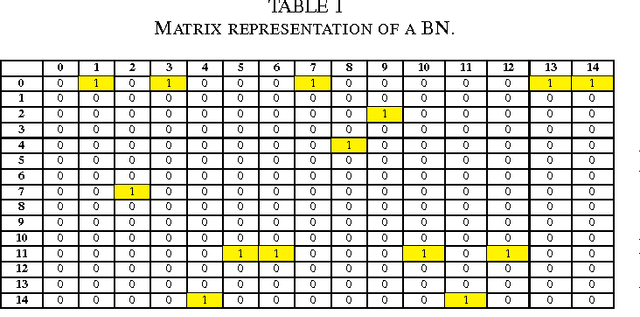
The emergence and development of cancer is a consequence of the accumulation over time of genomic mutations involving a specific set of genes, which provides the cancer clones with a functional selective advantage. In this work, we model the order of accumulation of such mutations during the progression, which eventually leads to the disease, by means of probabilistic graphic models, i.e., Bayesian Networks (BNs). We investigate how to perform the task of learning the structure of such BNs, according to experimental evidence, adopting a global optimization meta-heuristics. In particular, in this work we rely on Genetic Algorithms, and to strongly reduce the execution time of the inference -- which can also involve multiple repetitions to collect statistically significant assessments of the data -- we distribute the calculations using both multi-threading and a multi-node architecture. The results show that our approach is characterized by good accuracy and specificity; we also demonstrate its feasibility, thanks to a 84x reduction of the overall execution time with respect to a traditional sequential implementation.