 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEAD: an ensemble approach to detect adversarial examples from the hidden features of deep neural networks
Nov 25, 2021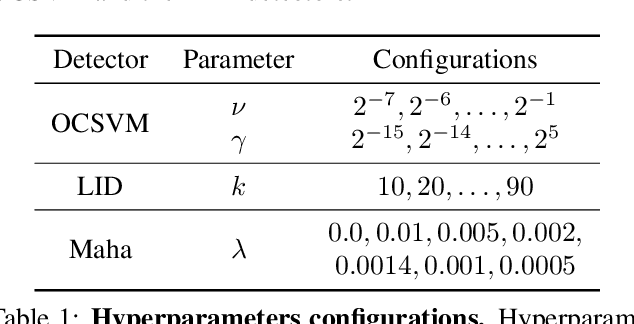
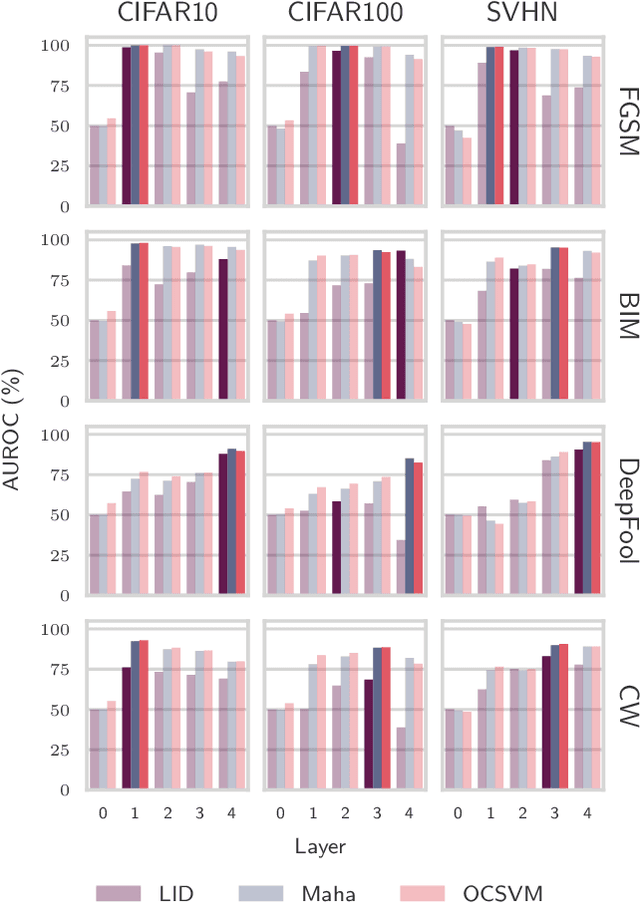
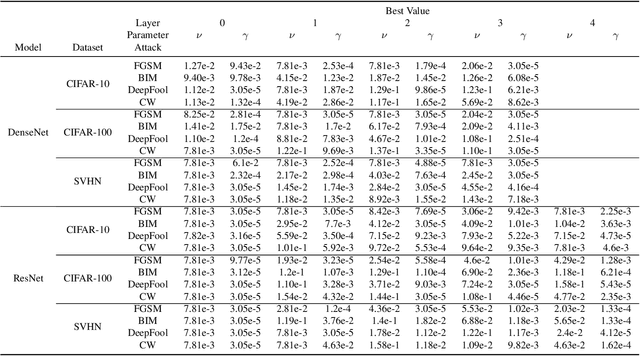
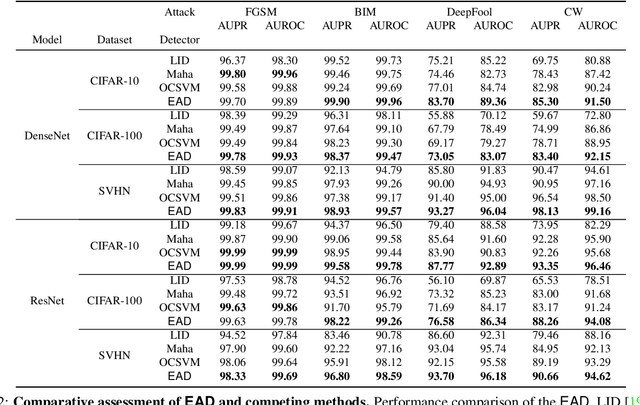
One of the key challenges in Deep Learning is the definition of effective strategies for the detection of adversarial examples. To this end, we propose a novel approach named Ensemble Adversarial Detector (EAD) for the identification of adversarial examples, in a standard multiclass classification scenario. EAD combines multiple detectors that exploit distinct properties of the input instances in the internal representation of a pre-trained Deep Neural Network (DNN). Specifically, EAD integrates the state-of-the-art detectors based on Mahalanobis distance and on Local Intrinsic Dimensionality (LID) with a newly introduced method based on One-class Support Vector Machines (OSVMs). Although all constituting methods assume that the greater the distance of a test instance from the set of correctly classified training instances, the higher its probability to be an adversarial example, they differ in the way such distance is computed. In order to exploit the effectiveness of the different methods in capturing distinct properties of data distributions and, accordingly, efficiently tackle the trade-off between generalization and overfitting, EAD employs detector-specific distance scores as features of a logistic regression classifier, after independent hyperparameters optimization. We evaluated the EAD approach on distinct datasets (CIFAR-10, CIFAR-100 and SVHN) and models (ResNet and DenseNet) and with regard to four adversarial attacks (FGSM, BIM, DeepFool and CW), also by comparing with competing approaches. Overall, we show that EAD achieves the best AUROC and AUPR in the large majority of the settings and comparable performance in the others. The improvement over the state-of-the-art, and the possibility to easily extend EAD to include any arbitrary set of detectors, pave the way to a widespread adoption of ensemble approaches in the broad field of adversarial example detection.
Investigating the Compositional Structure Of Deep Neural Networks
Feb 17, 2020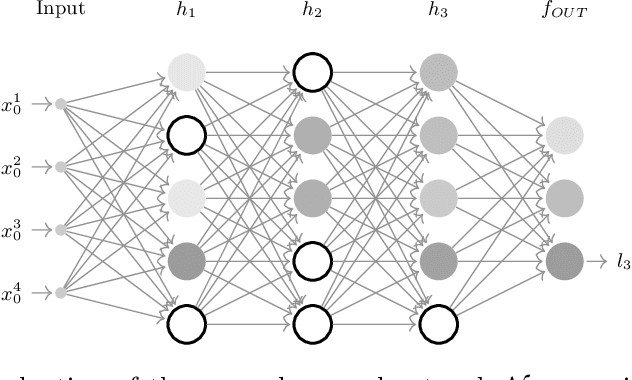
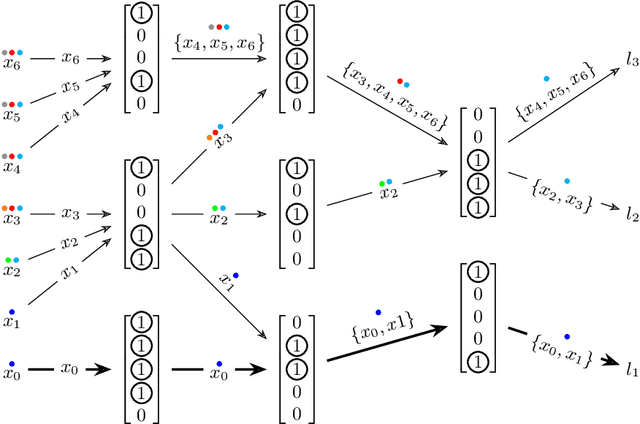
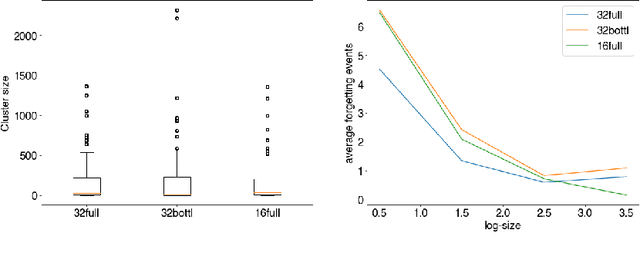
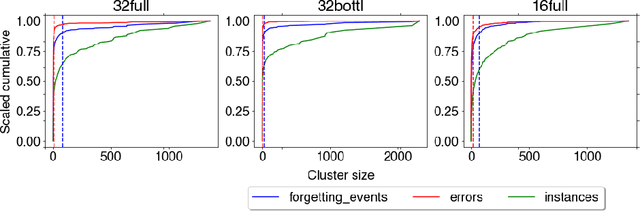
The current understanding of deep neural networks can only partially explain how input structure, network parameters and optimization algorithms jointly contribute to achieve the strong generalization power that is typically observed in many real-world applications. In order to improve the comprehension and interpretability of deep neural networks, we here introduce a novel theoretical framework based on the compositional structure of piecewise linear activation functions. By defining a direct acyclic graph representing the composition of activation patterns through the network layers, it is possible to characterize the instances of the input data with respect to both the predicted label and the specific (linear) transformation used to perform predictions. Preliminary tests on the MNIST dataset show that our method can group input instances with regard to their similarity in the internal representation of the neural network, providing an intuitive measure of input complexity.
Efficient computational strategies to learn the structure of probabilistic graphical models of cumulative phenomena
Oct 23, 2018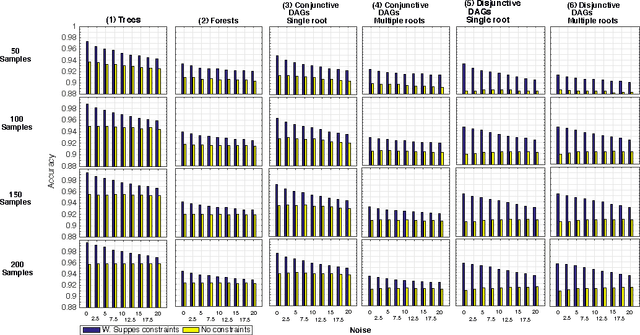
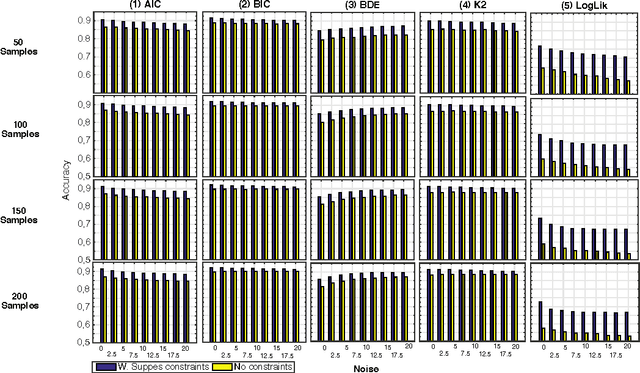
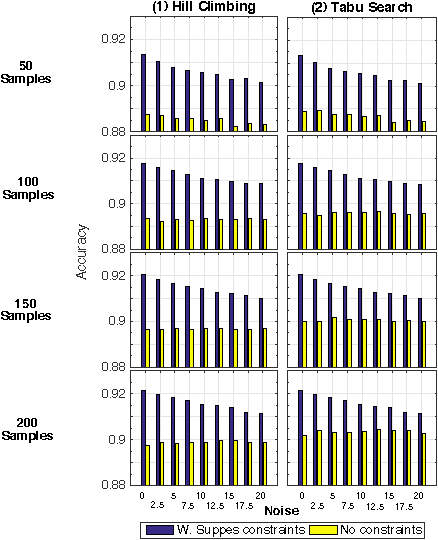
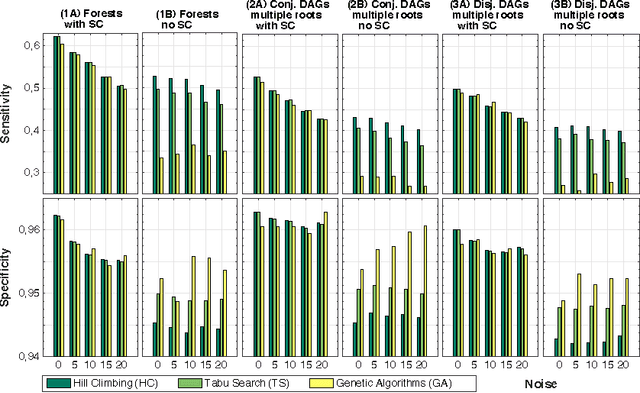
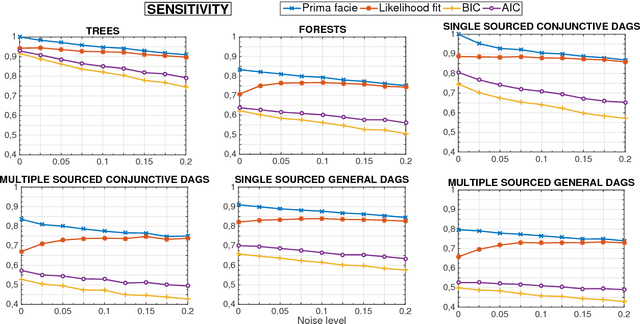
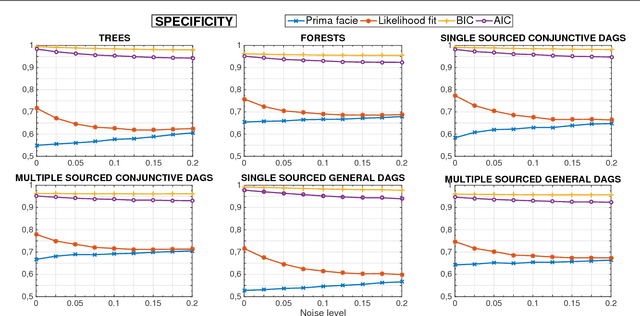
Structural learning of Bayesian Networks (BNs) is a NP-hard problem, which is further complicated by many theoretical issues, such as the I-equivalence among different structures. In this work, we focus on a specific subclass of BNs, named Suppes-Bayes Causal Networks (SBCNs), which include specific structural constraints based on Suppes' probabilistic causation to efficiently model cumulative phenomena. Here we compare the performance, via extensive simulations, of various state-of-the-art search strategies, such as local search techniques and Genetic Algorithms, as well as of distinct regularization methods. The assessment is performed on a large number of simulated datasets from topologies with distinct levels of complexity, various sample size and different rates of errors in the data. Among the main results, we show that the introduction of Suppes' constraints dramatically improve the inference accuracy, by reducing the solution space and providing a temporal ordering on the variables. We also report on trade-offs among different search techniques that can be efficiently employed in distinct experimental settings. This manuscript is an extended version of the paper "Structural Learning of Probabilistic Graphical Models of Cumulative Phenomena" presented at the 2018 International Conference on Computational Science.
Modeling cumulative biological phenomena with Suppes-Bayes Causal Networks
Jul 05, 2018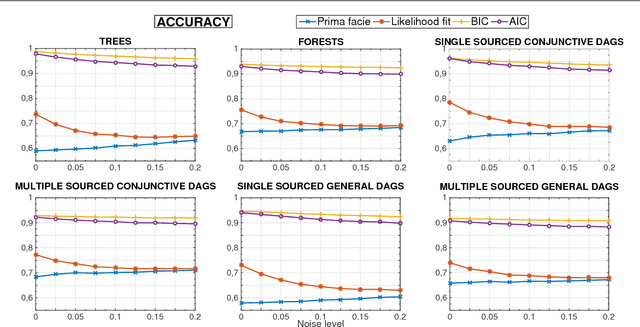

Several diseases related to cell proliferation are characterized by the accumulation of somatic DNA changes, with respect to wildtype conditions. Cancer and HIV are two common examples of such diseases, where the mutational load in the cancerous/viral population increases over time. In these cases, selective pressures are often observed along with competition, cooperation and parasitism among distinct cellular clones. Recently, we presented a mathematical framework to model these phenomena, based on a combination of Bayesian inference and Suppes' theory of probabilistic causation, depicted in graphical structures dubbed Suppes-Bayes Causal Networks (SBCNs). SBCNs are generative probabilistic graphical models that recapitulate the potential ordering of accumulation of such DNA changes during the progression of the disease. Such models can be inferred from data by exploiting likelihood-based model-selection strategies with regularization. In this paper we discuss the theoretical foundations of our approach and we investigate in depth the influence on the model-selection task of: (i) the poset based on Suppes' theory and (ii) different regularization strategies. Furthermore, we provide an example of application of our framework to HIV genetic data highlighting the valuable insights provided by the inferred.
Learning mutational graphs of individual tumor evolution from multi-sample sequencing data
Sep 04, 2017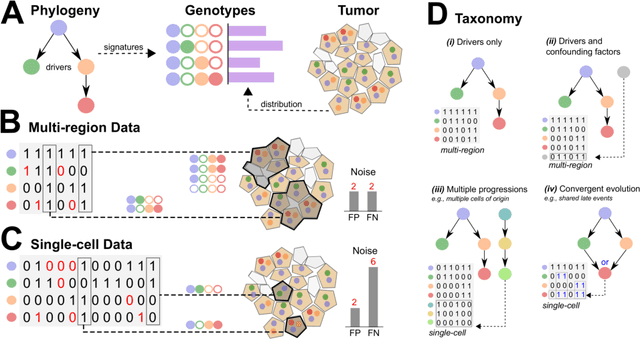
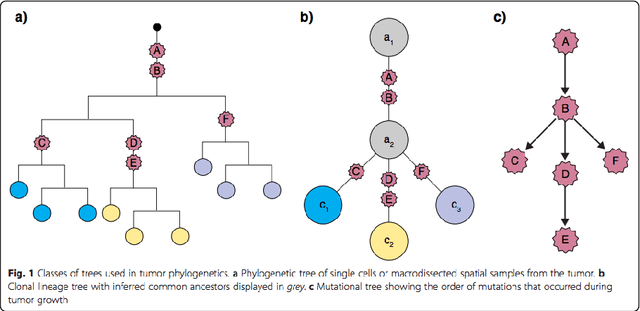
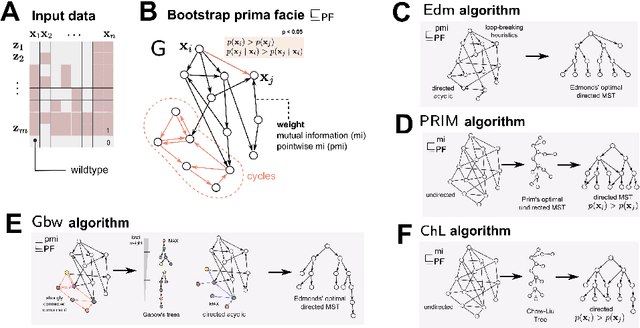
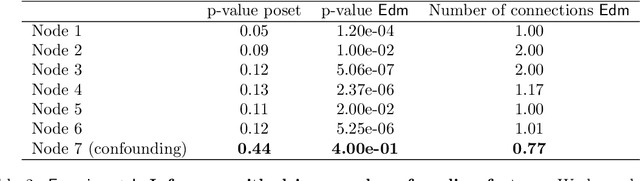
Phylogenetic techniques quantify intra-tumor heterogeneity by deconvolving either clonal or mutational trees from multi-sample sequencing data of individual tumors. Most of these methods rely on the well-known infinite sites assumption, and are limited to process either multi-region or single-cell sequencing data. Here, we improve over those methods with TRaIT, a unified statistical framework for the inference of the accumula- tion order of multiple types of genomic alterations driving tumor development. TRaIT supports both multi-region and single-cell sequencing data, and output mutational graphs accounting for violations of the infinite sites assumption due to convergent evolution, and other complex phenomena that cannot be detected with phylogenetic tools. Our method displays better accuracy, performance and robustness to noise and small sample size than state-of-the-art phylogenetic methods. We show with single-cell data from breast cancer and multi-region data from colorectal cancer that TRaIT can quantify the extent of intra-tumor heterogeneity and generate new testable experimental hypotheses.
Parallel Implementation of Efficient Search Schemes for the Inference of Cancer Progression Models
Mar 08, 2017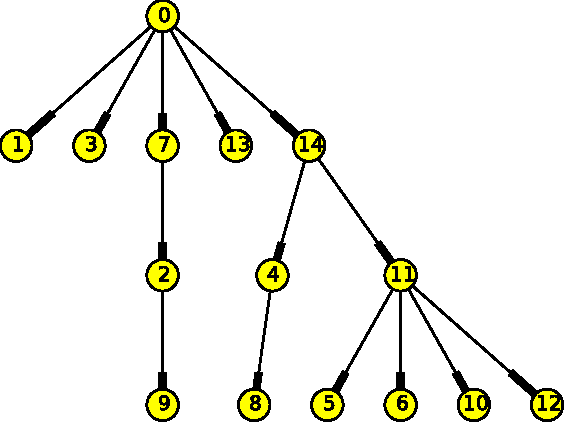
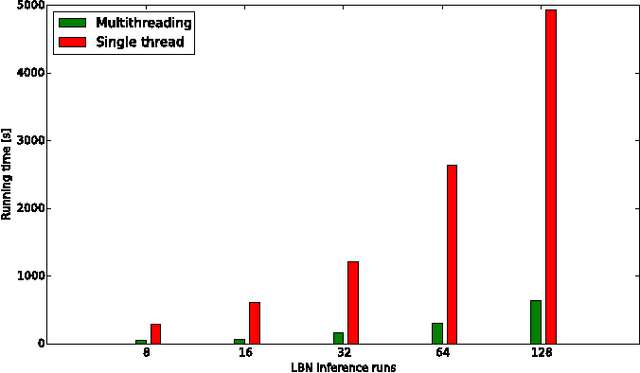
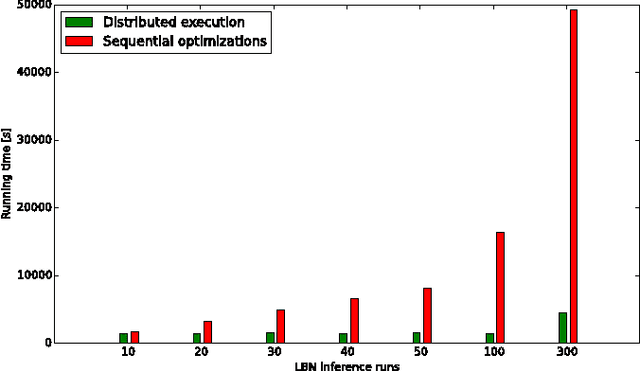
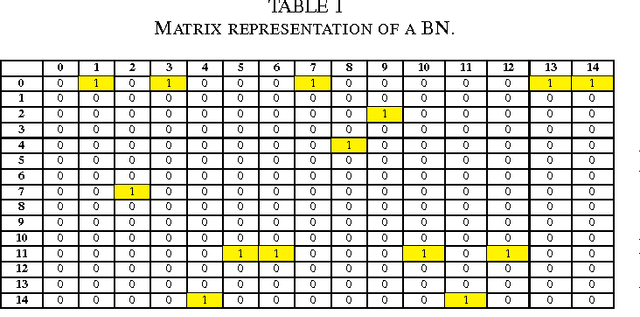
The emergence and development of cancer is a consequence of the accumulation over time of genomic mutations involving a specific set of genes, which provides the cancer clones with a functional selective advantage. In this work, we model the order of accumulation of such mutations during the progression, which eventually leads to the disease, by means of probabilistic graphic models, i.e., Bayesian Networks (BNs). We investigate how to perform the task of learning the structure of such BNs, according to experimental evidence, adopting a global optimization meta-heuristics. In particular, in this work we rely on Genetic Algorithms, and to strongly reduce the execution time of the inference -- which can also involve multiple repetitions to collect statistically significant assessments of the data -- we distribute the calculations using both multi-threading and a multi-node architecture. The results show that our approach is characterized by good accuracy and specificity; we also demonstrate its feasibility, thanks to a 84x reduction of the overall execution time with respect to a traditional sequential implementation.
Proceedings Wivace 2013 - Italian Workshop on Artificial Life and Evolutionary Computation
Sep 27, 2013The Wivace 2013 Electronic Proceedings in Theoretical Computer Science (EPTCS) contain some selected long and short articles accepted for the presentation at Wivace 2013 - Italian Workshop on Artificial Life and Evolutionary Computation, which was held at the University of Milan-Bicocca, Milan, on the 1st and 2nd of July, 2013.