 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeModeling cumulative biological phenomena with Suppes-Bayes Causal Networks
Jul 05, 2018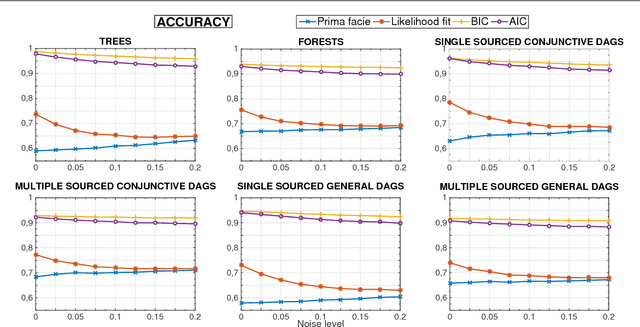
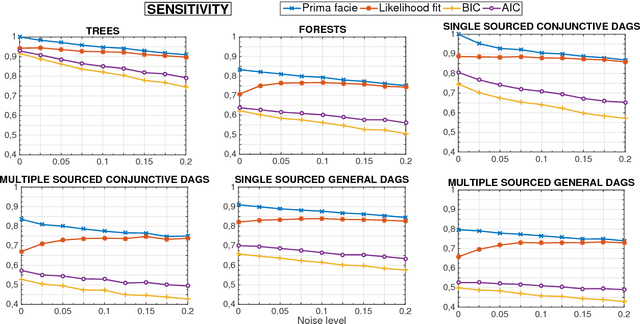
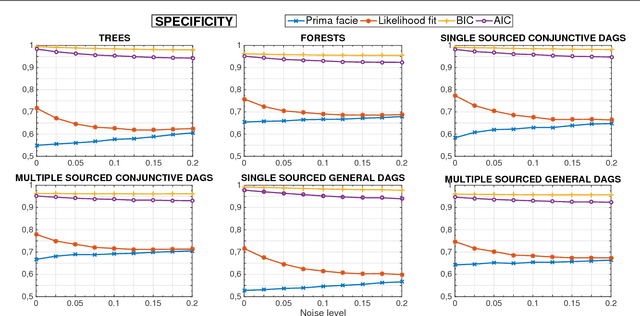

Several diseases related to cell proliferation are characterized by the accumulation of somatic DNA changes, with respect to wildtype conditions. Cancer and HIV are two common examples of such diseases, where the mutational load in the cancerous/viral population increases over time. In these cases, selective pressures are often observed along with competition, cooperation and parasitism among distinct cellular clones. Recently, we presented a mathematical framework to model these phenomena, based on a combination of Bayesian inference and Suppes' theory of probabilistic causation, depicted in graphical structures dubbed Suppes-Bayes Causal Networks (SBCNs). SBCNs are generative probabilistic graphical models that recapitulate the potential ordering of accumulation of such DNA changes during the progression of the disease. Such models can be inferred from data by exploiting likelihood-based model-selection strategies with regularization. In this paper we discuss the theoretical foundations of our approach and we investigate in depth the influence on the model-selection task of: (i) the poset based on Suppes' theory and (ii) different regularization strategies. Furthermore, we provide an example of application of our framework to HIV genetic data highlighting the valuable insights provided by the inferred.
Learning mutational graphs of individual tumor evolution from multi-sample sequencing data
Sep 04, 2017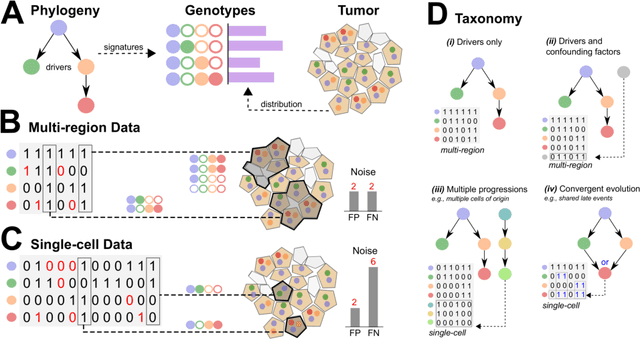
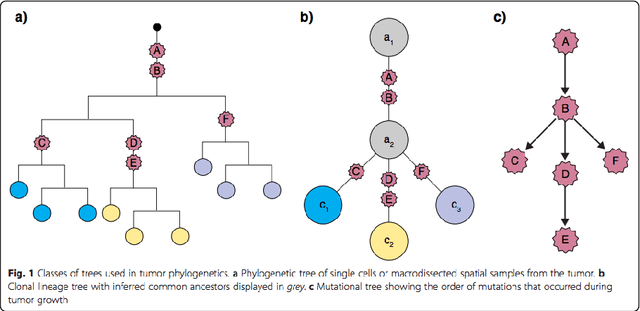
Phylogenetic techniques quantify intra-tumor heterogeneity by deconvolving either clonal or mutational trees from multi-sample sequencing data of individual tumors. Most of these methods rely on the well-known infinite sites assumption, and are limited to process either multi-region or single-cell sequencing data. Here, we improve over those methods with TRaIT, a unified statistical framework for the inference of the accumula- tion order of multiple types of genomic alterations driving tumor development. TRaIT supports both multi-region and single-cell sequencing data, and output mutational graphs accounting for violations of the infinite sites assumption due to convergent evolution, and other complex phenomena that cannot be detected with phylogenetic tools. Our method displays better accuracy, performance and robustness to noise and small sample size than state-of-the-art phylogenetic methods. We show with single-cell data from breast cancer and multi-region data from colorectal cancer that TRaIT can quantify the extent of intra-tumor heterogeneity and generate new testable experimental hypotheses.
On learning the structure of Bayesian Networks and submodular function maximization
Jun 07, 2017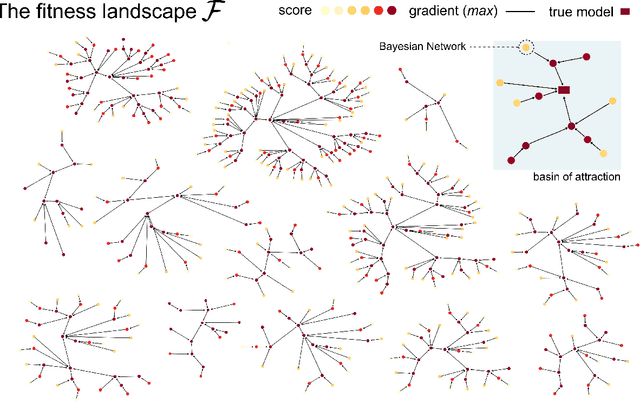
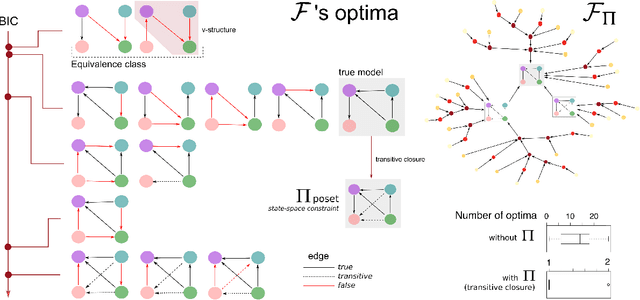
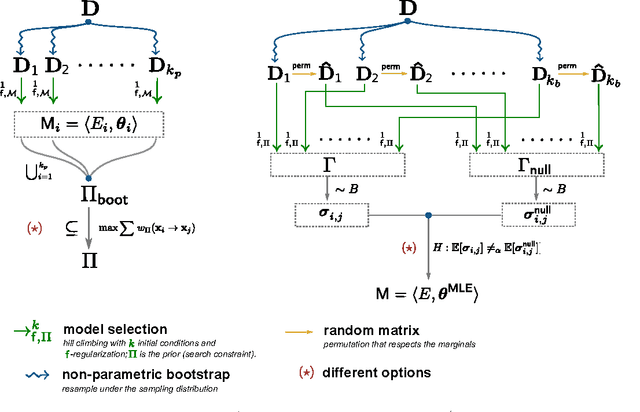
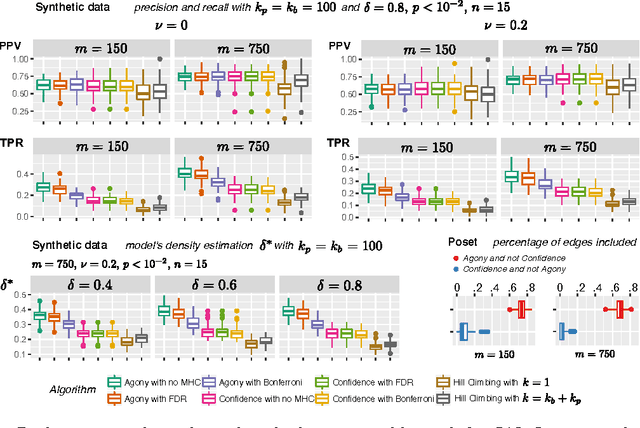
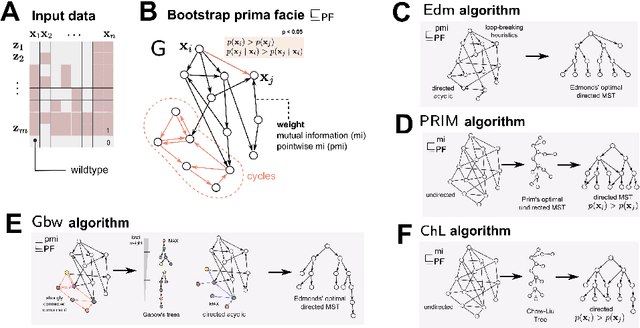
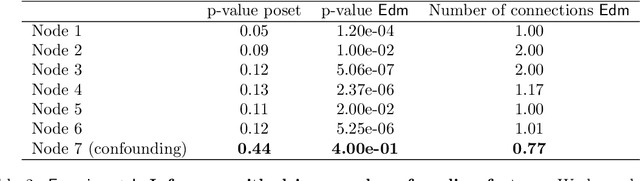
Learning the structure of dependencies among multiple random variables is a problem of considerable theoretical and practical interest. In practice, score optimisation with multiple restarts provides a practical and surprisingly successful solution, yet the conditions under which this may be a well founded strategy are poorly understood. In this paper, we prove that the problem of identifying the structure of a Bayesian Network via regularised score optimisation can be recast, in expectation, as a submodular optimisation problem, thus guaranteeing optimality with high probability. This result both explains the practical success of optimisation heuristics, and suggests a way to improve on such algorithms by artificially simulating multiple data sets via a bootstrap procedure. We show on several synthetic data sets that the resulting algorithm yields better recovery performance than the state of the art, and illustrate in a real cancer genomic study how such an approach can lead to valuable practical insights.
Matching models across abstraction levels with Gaussian Processes
May 07, 2016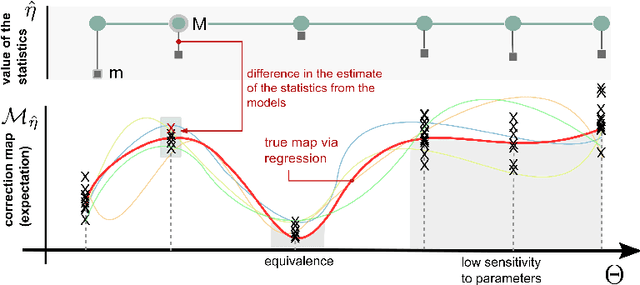
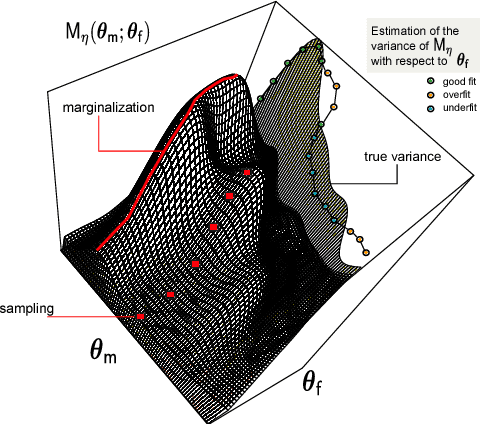
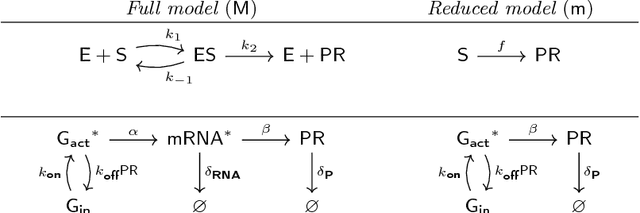
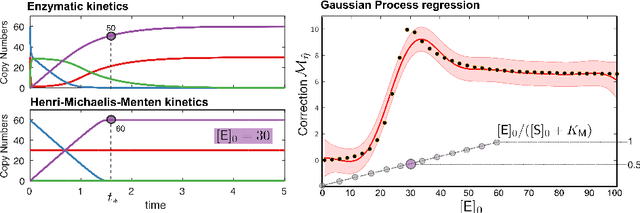
Biological systems are often modelled at different levels of abstraction depending on the particular aims/resources of a study. Such different models often provide qualitatively concordant predictions over specific parametrisations, but it is generally unclear whether model predictions are quantitatively in agreement, and whether such agreement holds for different parametrisations. Here we present a generally applicable statistical machine learning methodology to automatically reconcile the predictions of different models across abstraction levels. Our approach is based on defining a correction map, a random function which modifies the output of a model in order to match the statistics of the output of a different model of the same system. We use two biological examples to give a proof-of-principle demonstration of the methodology, and discuss its advantages and potential further applications.
Inference of Cancer Progression Models with Biological Noise
Aug 26, 2014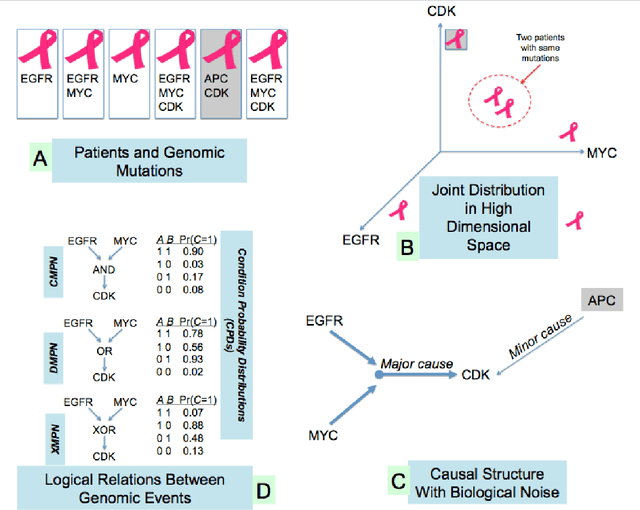
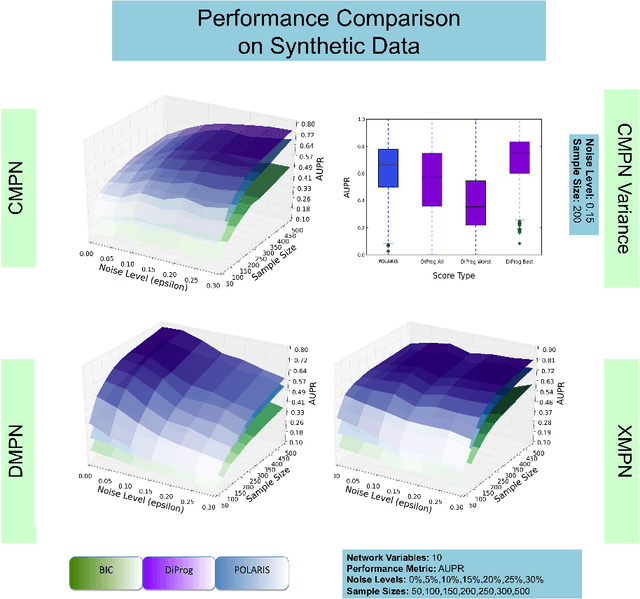
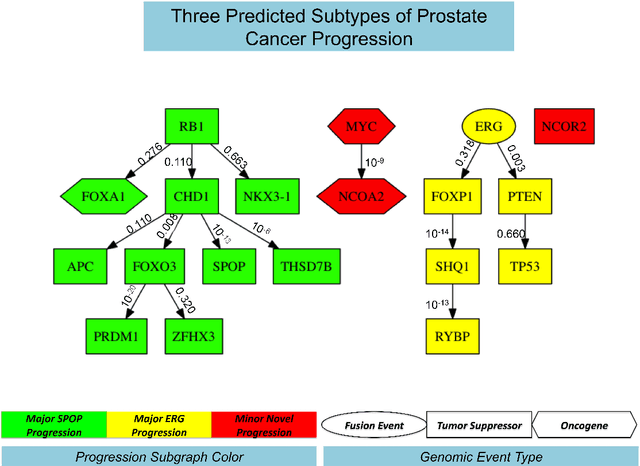
Many applications in translational medicine require the understanding of how diseases progress through the accumulation of persistent events. Specialized Bayesian networks called monotonic progression networks offer a statistical framework for modeling this sort of phenomenon. Current machine learning tools to reconstruct Bayesian networks from data are powerful but not suited to progression models. We combine the technological advances in machine learning with a rigorous philosophical theory of causation to produce Polaris, a scalable algorithm for learning progression networks that accounts for causal or biological noise as well as logical relations among genetic events, making the resulting models easy to interpret qualitatively. We tested Polaris on synthetically generated data and showed that it outperforms a widely used machine learning algorithm and approaches the performance of the competing special-purpose, albeit clairvoyant algorithm that is given a priori information about the model parameters. We also prove that under certain rather mild conditions, Polaris is guaranteed to converge for sufficiently large sample sizes. Finally, we applied Polaris to point mutation and copy number variation data in Prostate cancer from The Cancer Genome Atlas (TCGA) and found that there are likely three distinct progressions, one major androgen driven progression, one major non-androgen driven progression, and one novel minor androgen driven progression.
Proceedings Wivace 2013 - Italian Workshop on Artificial Life and Evolutionary Computation
Sep 27, 2013The Wivace 2013 Electronic Proceedings in Theoretical Computer Science (EPTCS) contain some selected long and short articles accepted for the presentation at Wivace 2013 - Italian Workshop on Artificial Life and Evolutionary Computation, which was held at the University of Milan-Bicocca, Milan, on the 1st and 2nd of July, 2013.