 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSVDq: 1.25-bit and 410x Key Cache Compression for LLM Attention
Feb 21, 2025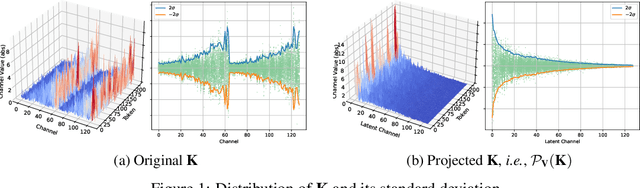
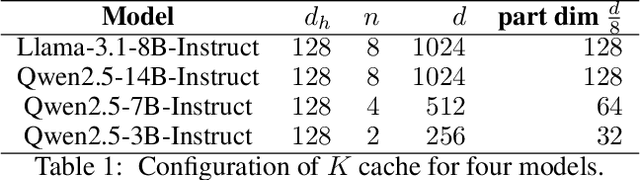
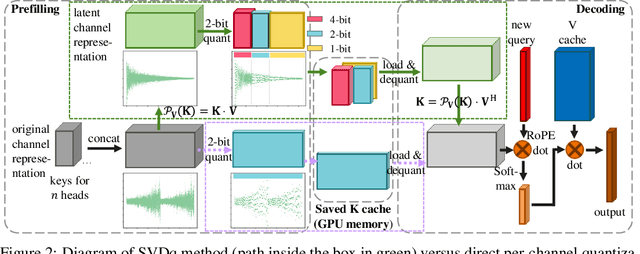
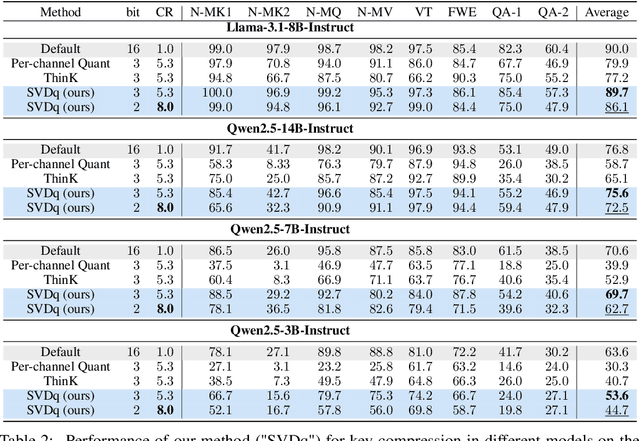
For the efficient inference of Large Language Models (LLMs), the effective compression of key-value (KV) cache is essential. Three main types of KV cache compression techniques, namely sparsity, channel compression, and quantization, have been identified. This study presents SVDq, a Singular Value Decomposition (SVD) - based mixed precision quantization method for K cache. Initially, K cache is transformed into latent channels using SVD basis representations. Since the values in latent channels decay rapidly and become negligible after only a few latent channels, our method then incorporates importance-aware quantization and compression for latent channels. This enables the effective allocation of higher precision to more significant channels. Theoretically, we prove that SVDq results in quantization errors (x0.1 or even lower) that are much lower than those of per-channel key quantization in the original space. Our findings based on RULER and LongBench benchmarks demonstrate that SVDq can achieve an equivalent key cache precision as low as 1.25-bit. When combined with key sparsity, it can reach a key compression ratio of up to 410x for attention computation, all while maintaining comparable model performance. Notably, our method is nearly lossless for LongBench datasets. This indicates that SVDq enables high-precision low-bit quantization, providing a more efficient solution for KV cache compression in LLMs.
Proxy-based Super Twisting Control Algorithm for Aerial Manipulators
May 06, 2023



Aerial manipulators are composed of an aerial multi-rotor that is equipped with a 6-DOF servo robot arm. To achieve precise position and attitude control during the arm's motion, it is critical for the system to have high performance control capabilities. However, the coupling effect between the multi-rotor UAVs' movement poses a challenge to the entire system's control capability. We have proposed a new proxy-based super twisting control approach for quadrotor UAVs that mitigates the disturbance caused by moving manipulators. This approach helps improve the stability of the aerial manipulation system when carrying out hovering or trajectory tracking tasks. The controller's effectiveness has been validated through numerical simulation and further tested in the Gazebo simulation environment.
Novel Modelling Strategies for High-frequency Stock Trading Data
Nov 30, 2022Full electronic automation in stock exchanges has recently become popular, generating high-frequency intraday data and motivating the development of near real-time price forecasting methods. Machine learning algorithms are widely applied to mid-price stock predictions. Processing raw data as inputs for prediction models (e.g., data thinning and feature engineering) can primarily affect the performance of the prediction methods. However, researchers rarely discuss this topic. This motivated us to propose three novel modelling strategies for processing raw data. We illustrate how our novel modelling strategies improve forecasting performance by analyzing high-frequency data of the Dow Jones 30 component stocks. In these experiments, our strategies often lead to statistically significant improvement in predictions. The three strategies improve the F1 scores of the SVM models by 0.056, 0.087, and 0.016, respectively.
A systematic evaluation of methods for cell phenotype classification using single-cell RNA sequencing data
Oct 01, 2021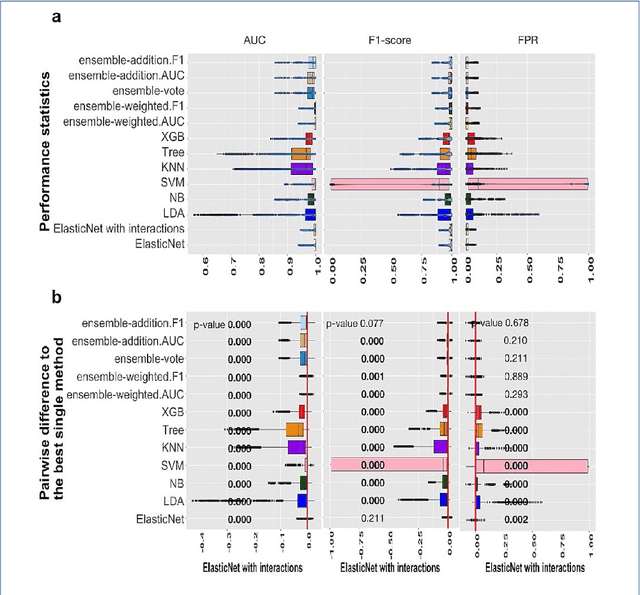
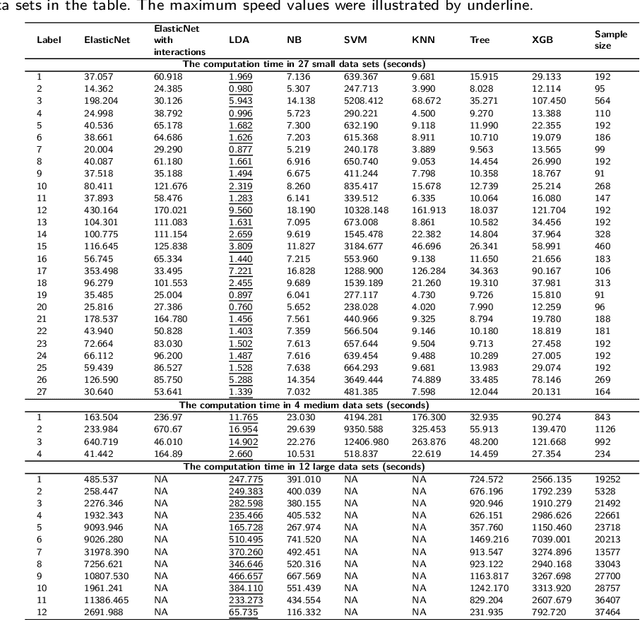
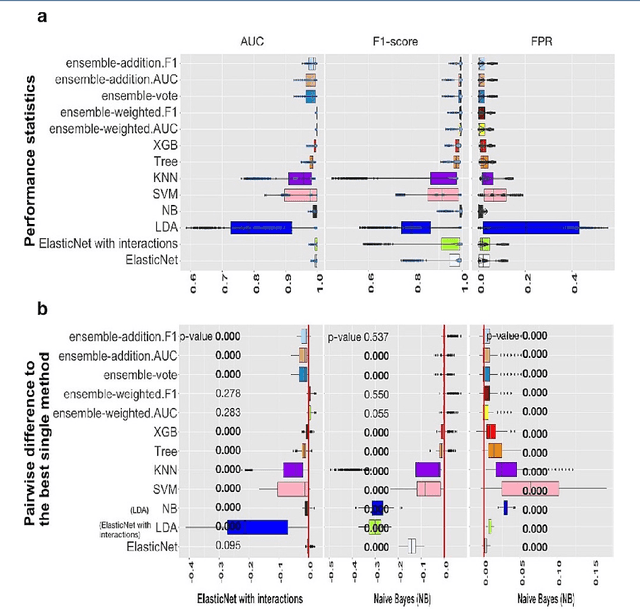
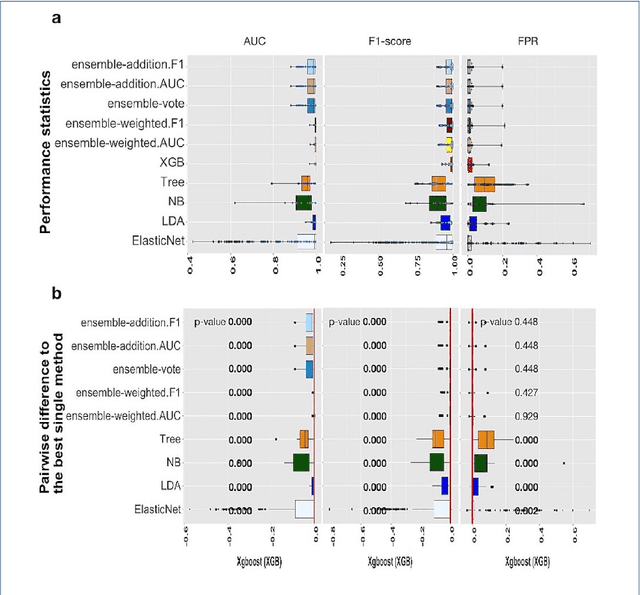
Background: Single-cell RNA sequencing (scRNA-seq) yields valuable insights about gene expression and gives critical information about complex tissue cellular composition. In the analysis of single-cell RNA sequencing, the annotations of cell subtypes are often done manually, which is time-consuming and irreproducible. Garnett is a cell-type annotation software based the on elastic net method. Besides cell-type annotation, supervised machine learning methods can also be applied to predict other cell phenotypes from genomic data. Despite the popularity of such applications, there is no existing study to systematically investigate the performance of those supervised algorithms in various sizes of scRNA-seq data sets. Methods and Results: This study evaluates 13 popular supervised machine learning algorithms to classify cell phenotypes, using published real and simulated data sets with diverse cell sizes. The benchmark contained two parts. In the first part, we used real data sets to assess the popular supervised algorithms' computing speed and cell phenotype classification performance. The classification performances were evaluated using AUC statistics, F1-score, precision, recall, and false-positive rate. In the second part, we evaluated gene selection performance using published simulated data sets with a known list of real genes. Conclusion: The study outcomes showed that ElasticNet with interactions performed best in small and medium data sets. NB was another appropriate method for medium data sets. In large data sets, XGB works excellent. Ensemble algorithms were not significantly superior to individual machine learning methods. Adding interactions to ElasticNet can help, and the improvement was significant in small data sets.
Multi-Stage Graph Peeling Algorithm for Probabilistic Core Decomposition
Aug 13, 2021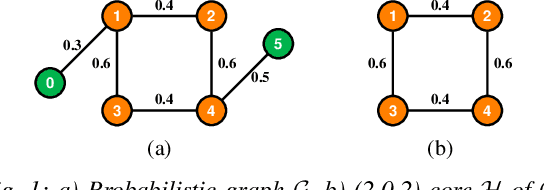

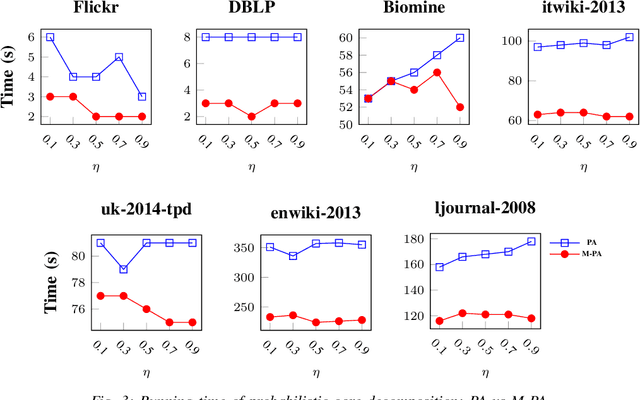
Mining dense subgraphs where vertices connect closely with each other is a common task when analyzing graphs. A very popular notion in subgraph analysis is core decomposition. Recently, Esfahani et al. presented a probabilistic core decomposition algorithm based on graph peeling and Central Limit Theorem (CLT) that is capable of handling very large graphs. Their proposed peeling algorithm (PA) starts from the lowest degree vertices and recursively deletes these vertices, assigning core numbers, and updating the degree of neighbour vertices until it reached the maximum core. However, in many applications, particularly in biology, more valuable information can be obtained from dense sub-communities and we are not interested in small cores where vertices do not interact much with others. To make the previous PA focus more on dense subgraphs, we propose a multi-stage graph peeling algorithm (M-PA) that has a two-stage data screening procedure added before the previous PA. After removing vertices from the graph based on the user-defined thresholds, we can reduce the graph complexity largely and without affecting the vertices in subgraphs that we are interested in. We show that M-PA is more efficient than the previous PA and with the properly set filtering threshold, can produce very similar if not identical dense subgraphs to the previous PA (in terms of graph density and clustering coefficient).
Handling highly correlated genes of Single-Cell RNA sequencing data in prediction models
Jul 05, 2020

Motivation: Selecting feature genes and predicting cells' phenotype are typical tasks in the analysis of scRNA-seq data. Many algorithms were developed for these tasks, but high correlations among genes create challenges specifically in scRNA-seq analysis, which are not well addressed. Highly correlated genes lead to collinearity and unreliable model fitting. Highly correlated genes compete with each other in feature selection, which causes underestimation of their importance. Most importantly, when a causal gene is highly correlated other genes, most algorithms select one of them in a data driven manner. The correlation structure among genes could change substantially. Hence, it is critical to build a prediction model based on causal genes but not their highly correlated genes. Results: To address the issues discussed above, we propose a grouping algorithm which can be integrated in prediction models. Using real benchmark scRNA-seq data sets and simulated cell phenotypes, we show our novel method significantly outperform standard prediction models in the performance of both prediction and feature selection. Our algorithm report the whole group of correlated genes, which allow researchers to conduct additional studies to identify the causal genes from the group. Availability: An R package is being developed and will be made available on the Comprehensive R Archive Network (CRAN). In the meantime, R code can be requested by email.
Simultaneous prediction of multiple outcomes using revised stacking algorithms
Jan 29, 2019
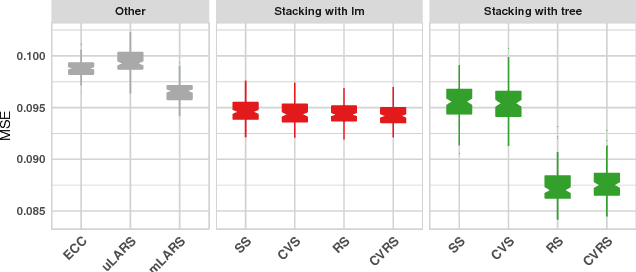

Motivation: HIV is difficult to treat because its virus mutates at a high rate and mutated viruses easily develop resistance to existing drugs. If the relationships between mutations and drug resistances can be determined from historical data, patients can be provided personalized treatment according to their own mutation information. The HIV Drug Resistance Database was built to investigate the relationships. Our goal is to build a model using data in this database, which simultaneously predicts the resistance of multiple drugs using mutation information from sequences of viruses for any new patient. Results: We propose two variations of a stacking algorithm which borrow information among multiple prediction tasks to improve multivariate prediction performance. The most attractive feature of our proposed methods is the flexibility with which complex multivariate prediction models can be constructed using any univariate prediction models. Using cross-validation studies, we show that our proposed methods outperform other popular multivariate prediction methods. Availability: An R package will be made available.
Bayesian hierarchical models for SNP discovery from genome-wide association studies, a semi-supervised machine learning approach
Jun 21, 2018Genome-wide association studies (GWASs) aim to detect genetic risk factors for complex human diseases by identifying disease-associated single-nucleotide polymorphisms (SNPs). SNP-wise approach, the standard method for analyzing GWAS, tests each SNP individually. Then the P-values are adjusted for multiple testing. Multiple testing adjustment (purely based on p-values) is over-conservative and causes lack of power in many GWASs, due to insufficiently modelling the relationship among SNPs. To address this problem, we propose a novel method, which borrows information across SNPs by grouping SNPs into three clusters. We pre-specify the patterns of clusters by minor allele frequencies of SNPs between cases and controls, and enforce the patterns with prior distributions. Therefore, compared with the traditional approach, it better controls false discovery rate (FDR) and shows higher sensitivity, which is confirmed by our simulation studies. We re-analyzed real data studies on identifying SNPs associated with severe bortezomib-induced peripheral neuropathy (BiPN) in patients with multiple myeloma. The original analysis in the literature failed to identify SNPs after FDR adjustment. Our proposed method not only detected the reported SNPs after FDR adjustment but also discovered a novel SNP rs4351714 that has been reported to be related to multiple myeloma in another study.