 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHandling highly correlated genes of Single-Cell RNA sequencing data in prediction models
Jul 05, 2020

Motivation: Selecting feature genes and predicting cells' phenotype are typical tasks in the analysis of scRNA-seq data. Many algorithms were developed for these tasks, but high correlations among genes create challenges specifically in scRNA-seq analysis, which are not well addressed. Highly correlated genes lead to collinearity and unreliable model fitting. Highly correlated genes compete with each other in feature selection, which causes underestimation of their importance. Most importantly, when a causal gene is highly correlated other genes, most algorithms select one of them in a data driven manner. The correlation structure among genes could change substantially. Hence, it is critical to build a prediction model based on causal genes but not their highly correlated genes. Results: To address the issues discussed above, we propose a grouping algorithm which can be integrated in prediction models. Using real benchmark scRNA-seq data sets and simulated cell phenotypes, we show our novel method significantly outperform standard prediction models in the performance of both prediction and feature selection. Our algorithm report the whole group of correlated genes, which allow researchers to conduct additional studies to identify the causal genes from the group. Availability: An R package is being developed and will be made available on the Comprehensive R Archive Network (CRAN). In the meantime, R code can be requested by email.
Simultaneous prediction of multiple outcomes using revised stacking algorithms
Jan 29, 2019
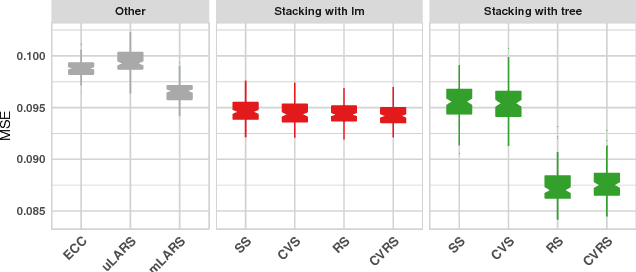

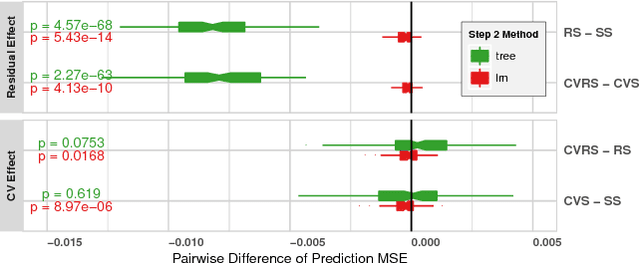
Motivation: HIV is difficult to treat because its virus mutates at a high rate and mutated viruses easily develop resistance to existing drugs. If the relationships between mutations and drug resistances can be determined from historical data, patients can be provided personalized treatment according to their own mutation information. The HIV Drug Resistance Database was built to investigate the relationships. Our goal is to build a model using data in this database, which simultaneously predicts the resistance of multiple drugs using mutation information from sequences of viruses for any new patient. Results: We propose two variations of a stacking algorithm which borrow information among multiple prediction tasks to improve multivariate prediction performance. The most attractive feature of our proposed methods is the flexibility with which complex multivariate prediction models can be constructed using any univariate prediction models. Using cross-validation studies, we show that our proposed methods outperform other popular multivariate prediction methods. Availability: An R package will be made available.
A Bayesian Group Sparse Multi-Task Regression Model for Imaging Genetics
Oct 17, 2016
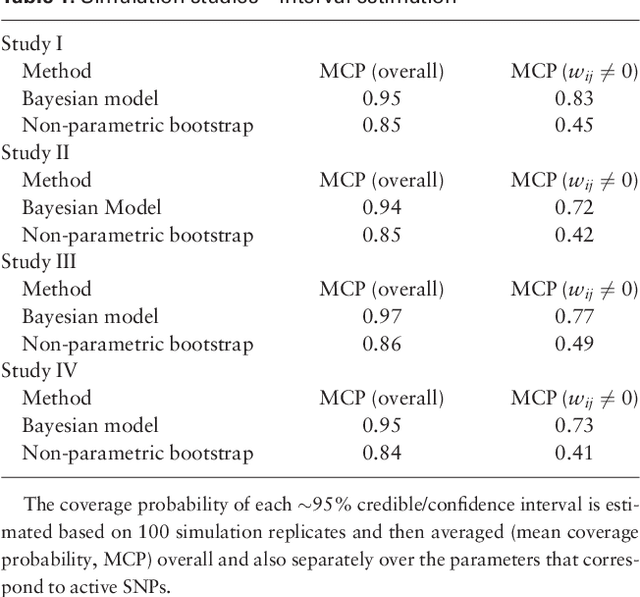
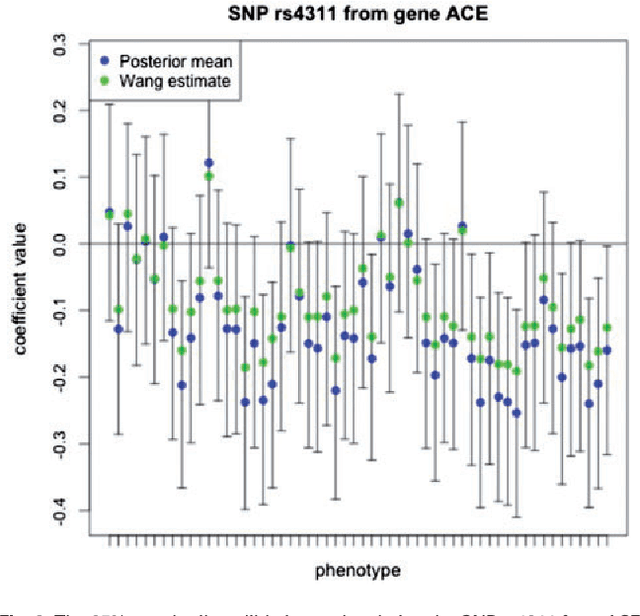
Motivation: Recent advances in technology for brain imaging and high-throughput genotyping have motivated studies examining the influence of genetic variation on brain structure. Wang et al. (Bioinformatics, 2012) have developed an approach for the analysis of imaging genomic studies using penalized multi-task regression with regularization based on a novel group $l_{2,1}$-norm penalty which encourages structured sparsity at both the gene level and SNP level. While incorporating a number of useful features, the proposed method only furnishes a point estimate of the regression coefficients; techniques for conducting statistical inference are not provided. A new Bayesian method is proposed here to overcome this limitation. Results: We develop a Bayesian hierarchical modeling formulation where the posterior mode corresponds to the estimator proposed by Wang et al. (Bioinformatics, 2012), and an approach that allows for full posterior inference including the construction of interval estimates for the regression parameters. We show that the proposed hierarchical model can be expressed as a three-level Gaussian scale mixture and this representation facilitates the use of a Gibbs sampling algorithm for posterior simulation. Simulation studies demonstrate that the interval estimates obtained using our approach achieve adequate coverage probabilities that outperform those obtained from the nonparametric bootstrap. Our proposed methodology is applied to the analysis of neuroimaging and genetic data collected as part of the Alzheimer's Disease Neuroimaging Initiative (ADNI), and this analysis of the ADNI cohort demonstrates clearly the value added of incorporating interval estimation beyond only point estimation when relating SNPs to brain imaging endophenotypes.
Regularization Parameter Selection for a Bayesian Multi-Level Group Lasso Regression Model with Application to Imaging Genomics
Mar 27, 2016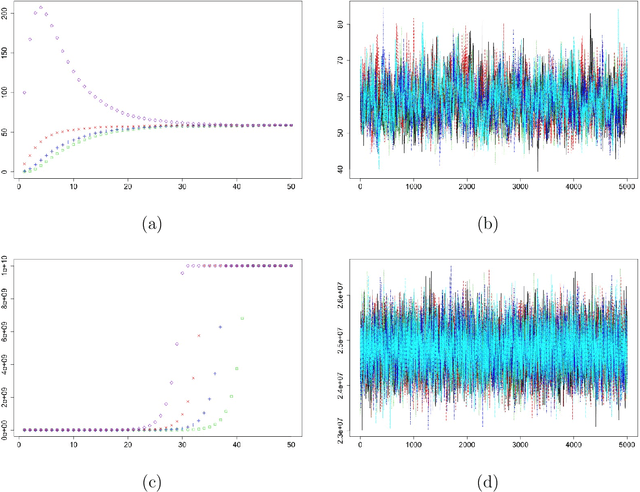
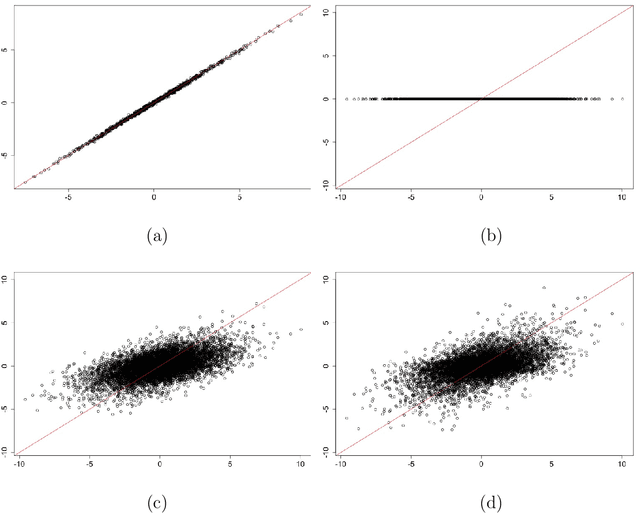
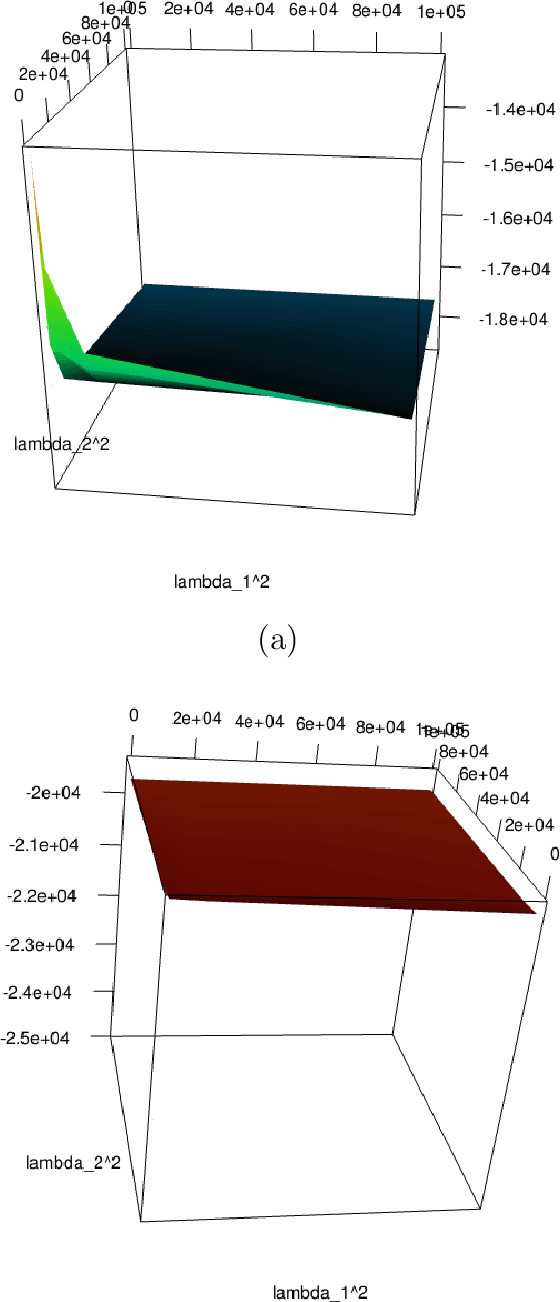
We investigate the choice of tuning parameters for a Bayesian multi-level group lasso model developed for the joint analysis of neuroimaging and genetic data. The regression model we consider relates multivariate phenotypes consisting of brain summary measures (volumetric and cortical thickness values) to single nucleotide polymorphism (SNPs) data and imposes penalization at two nested levels, the first corresponding to genes and the second corresponding to SNPs. Associated with each level in the penalty is a tuning parameter which corresponds to a hyperparameter in the hierarchical Bayesian formulation. Following previous work on Bayesian lassos we consider the estimation of tuning parameters through either hierarchical Bayes based on hyperpriors and Gibbs sampling or through empirical Bayes based on maximizing the marginal likelihood using a Monte Carlo EM algorithm. For the specific model under consideration we find that these approaches can lead to severe overshrinkage of the regression parameter estimates in the high-dimensional setting or when the genetic effects are weak. We demonstrate these problems through simulation examples and study an approximation to the marginal likelihood which sheds light on the cause of this problem. We then suggest an alternative approach based on the widely applicable information criterion (WAIC), an asymptotic approximation to leave-one-out cross-validation that can be computed conveniently within an MCMC framework.