 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAn Improved Adaptive PID Optimizer with Enhanced Convergence and Stability for Deep Learning
May 21, 2026Optimization is essential in deep learning. The foundational method upon which most optimizers are built is momentum-based stochastic gradient descent. However, it suffers from two key drawbacks. First, it has noisy and varying gradients, and second, it has an overshoot phenomenon. To address noisy gradients, Adam was proposed, which remains the most widely used adaptive optimizer. To address the overshoot phenomenon, a control-theory-based PID optimizer was proposed. To tackle both the limitations within a single framework, several variants of Adaptive PID (AdaPID) have recently been proposed. Although AdaPID performs well, it still inherits two critical drawbacks from Adam, namely convergence and stability issues. In this work, we address both these limitations. To fix the convergence issue, we uniquely integrate the idea of using a non-increasing effective learning rate into AdaPID (originally proposed in AMSGrad, an extension of Adam). To fix the stability issue, we innovatively integrate a gradient difference based modulation factor into AdaPID (originally proposed in DiffGrad, another extension of Adam). Combining both these ideas in AdaPID, results in our novel IAdaPID-ADG optimizer. We evaluate our proposed optimizer on multiple datasets, including benchmark datasets (MNIST and CIFAR10) and real-world datasets (IARC and AnnoCerv). The IAdaPID-ADG substantially outperforms all competing optimizers. Additionally, we perform an ablation study on the MNIST dataset to demonstrate the contribution of each added component.
ResGene-T: A Tensor-Based Residual Network Approach for Genomic Prediction
Feb 28, 2026In this work, we propose a new deep learning model for Genomic Prediction (GP), which involves correlating genotypic data with phenotypic. The genotypes are typically fed as a sequence of characters to the 1D-Convolution Neural Network layer of the underlying deep learning model. Inspired by earlier work that represented genotype as a 2D-image for genotype-phenotype classification, we extend this idea to GP, which is a regression task. We use a ResNet-18 as the underlying architecture, and term this model as ResGene-2D. Although the 2D-image representation captures biological interactions well, it requires all the layers of the model to do so. This limits training efficiency. Thus, as seen in the earlier work that proposed a 2D-image representation, our ResGene-2D performs almost the same as other models (3% improvement). To overcome this, we propose a novel idea of converting the 2D-image into a 3D/ tensor and feed this to the ResNet-18 architecture, and term this model as ResGene-T. We evaluate our proposed models on three crop species having ten phenotypic traits and compare it with seven most popular models (two statistical, two machine learning, and three deep learning). ResGene-T performs the best among all these seven methods (gains from 14.51% to 41.51%).
Chameleon2++: An Efficient Chameleon2 Clustering with Approximate Nearest Neighbors
Jan 05, 2025Clustering algorithms are fundamental tools in data analysis, with hierarchical methods being particularly valuable for their flexibility. Chameleon is a widely used hierarchical clustering algorithm that excels at identifying high-quality clusters of arbitrary shapes, sizes, and densities. Chameleon2 is the most recent variant that has demonstrated significant improvements, but suffers from critical failings and there are certain improvements that can be made. The first failure we address is that the complexity of Chameleon2 is claimed to be $O(n^2)$, while we demonstrate that it is actually $O(n^2\log{n})$, with $n$ being the number of data points. Furthermore, we suggest improvements to Chameleon2 that ensure that the complexity remains $O(n^2)$ with minimal to no loss of performance. The second failing of Chameleon2 is that it lacks transparency and it does not provide the fine-tuned algorithm parameters used to obtain the claimed results. We meticulously provide all such parameter values to enhance replicability. The improvement which we make in Chameleon2 is that we replace the exact $k$-NN search with an approximate $k$-NN search. This further reduces the algorithmic complexity down to $O(n\log{n})$ without any performance loss. Here, we primarily configure three approximate nearest neighbor search algorithms (Annoy, FLANN and NMSLIB) to align with the overarching Chameleon2 clustering framework. Experimental evaluations on standard benchmark datasets demonstrate that the proposed Chameleon2++ algorithm is more efficient, robust, and computationally optimal.
AI Algorithm for Predicting and Optimizing Trajectory of UAV Swarm
May 20, 2024This paper explores the application of Artificial Intelligence (AI) techniques for generating the trajectories of fleets of Unmanned Aerial Vehicles (UAVs). The two main challenges addressed include accurately predicting the paths of UAVs and efficiently avoiding collisions between them. Firstly, the paper systematically applies a diverse set of activation functions to a Feedforward Neural Network (FFNN) with a single hidden layer, which enhances the accuracy of the predicted path compared to previous work. Secondly, we introduce a novel activation function, AdaptoSwelliGauss, which is a sophisticated fusion of Swish and Elliott activations, seamlessly integrated with a scaled and shifted Gaussian component. Swish facilitates smooth transitions, Elliott captures abrupt trajectory changes, and the scaled and shifted Gaussian enhances robustness against noise. This dynamic combination is specifically designed to excel in capturing the complexities of UAV trajectory prediction. This new activation function gives substantially better accuracy than all existing activation functions. Thirdly, we propose a novel Integrated Collision Detection, Avoidance, and Batching (ICDAB) strategy that merges two complementary UAV collision avoidance techniques: changing UAV trajectories and altering their starting times, also referred to as batching. This integration helps overcome the disadvantages of both - reduction in the number of trajectory manipulations, which avoids overly convoluted paths in the first technique, and smaller batch sizes, which reduce overall takeoff time in the second.
Deep Learning Descriptor Hybridization with Feature Reduction for Accurate Cervical Cancer Colposcopy Image Classification
May 01, 2024Cervical cancer stands as a predominant cause of female mortality, underscoring the need for regular screenings to enable early diagnosis and preemptive treatment of pre-cancerous conditions. The transformation zone in the cervix, where cellular differentiation occurs, plays a critical role in the detection of abnormalities. Colposcopy has emerged as a pivotal tool in cervical cancer prevention since it provides a meticulous examination of cervical abnormalities. However, challenges in visual evaluation necessitate the development of Computer Aided Diagnosis (CAD) systems. We propose a novel CAD system that combines the strengths of various deep-learning descriptors (ResNet50, ResNet101, and ResNet152) with appropriate feature normalization (min-max) as well as feature reduction technique (LDA). The combination of different descriptors ensures that all the features (low-level like edges and colour, high-level like shape and texture) are captured, feature normalization prevents biased learning, and feature reduction avoids overfitting. We do experiments on the IARC dataset provided by WHO. The dataset is initially segmented and balanced. Our approach achieves exceptional performance in the range of 97%-100% for both the normal-abnormal and the type classification. A competitive approach for type classification on the same dataset achieved 81%-91% performance.
A Novel Sampled Clustering Algorithm for Rice Phenotypic Data
Dec 22, 2023Phenotypic (or Physical) characteristics of plant species are commonly used to perform clustering. In one of our recent works (Shastri et al. (2021)), we used a probabilistically sampled (using pivotal sampling) and spectrally clustered algorithm to group soybean species. These techniques were used to obtain highly accurate clusterings at a reduced cost. In this work, we extend the earlier algorithm to cluster rice species. We improve the base algorithm in three ways. First, we propose a new function to build the similarity matrix in Spectral Clustering. Commonly, a natural exponential function is used for this purpose. Based upon the spectral graph theory and the involved Cheeger's inequality, we propose the use a base "a" exponential function instead. This gives a similarity matrix spectrum favorable for clustering, which we support via an eigenvalue analysis. Second, the function used to build the similarity matrix in Spectral Clustering was earlier scaled with a fixed factor (called global scaling). Based upon the idea of Zelnik-Manor and Perona (2004), we now use a factor that varies with matrix elements (called local scaling) and works better. Third, to compute the inclusion probability of a specie in the pivotal sampling algorithm, we had earlier used the notion of deviation that captured how far specie's characteristic values were from their respective base values (computed over all species). A maximum function was used before to find the base values. We now use a median function, which is more intuitive. We support this choice using a statistical analysis. With experiments on 1865 rice species, we demonstrate that in terms of silhouette values, our new Sampled Spectral Clustering is 61% better than Hierarchical Clustering (currently prevalent). Also, our new algorithm is significantly faster than Hierarchical Clustering due to the involved sampling.
Cube Sampled K-Prototype Clustering for Featured Data
Aug 23, 2021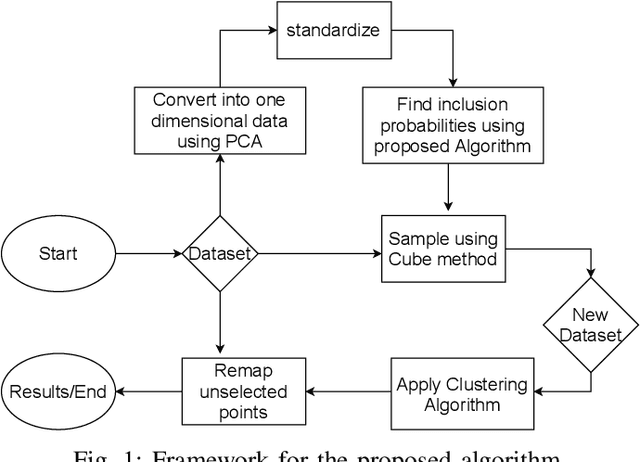
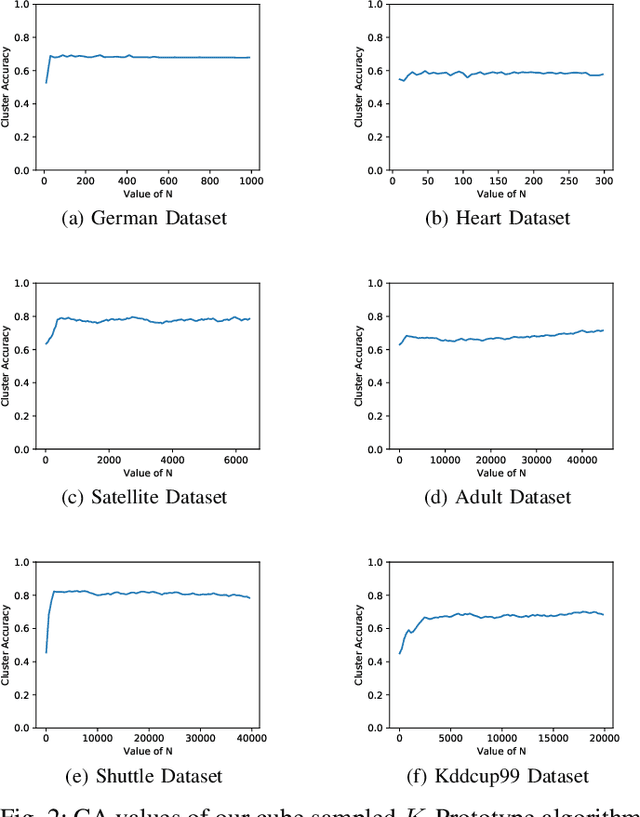
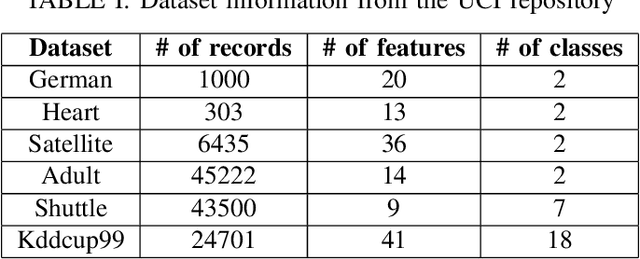
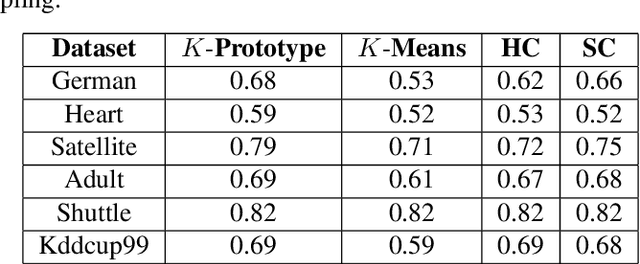
Clustering large amount of data is becoming increasingly important in the current times. Due to the large sizes of data, clustering algorithm often take too much time. Sampling this data before clustering is commonly used to reduce this time. In this work, we propose a probabilistic sampling technique called cube sampling along with K-Prototype clustering. Cube sampling is used because of its accurate sample selection. K-Prototype is most frequently used clustering algorithm when the data is numerical as well as categorical (very common in today's time). The novelty of this work is in obtaining the crucial inclusion probabilities for cube sampling using Principal Component Analysis (PCA). Experiments on multiple datasets from the UCI repository demonstrate that cube sampled K-Prototype algorithm gives the best clustering accuracy among similarly sampled other popular clustering algorithms (K-Means, Hierarchical Clustering (HC), Spectral Clustering (SC)). When compared with unsampled K-Prototype, K-Means, HC and SC, it still has the best accuracy with the added advantage of reduced computational complexity (due to reduced data size).
Probabilistically Sampled and Spectrally Clustered Plant Genotypes using Phenotypic Characteristics
Sep 18, 2020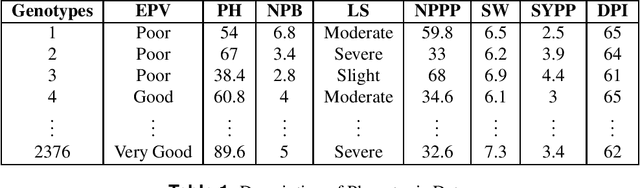
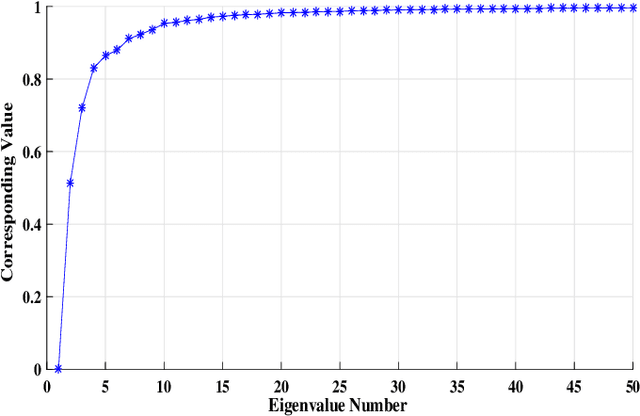
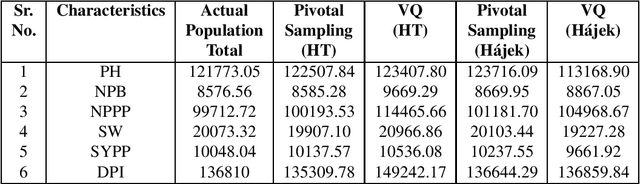
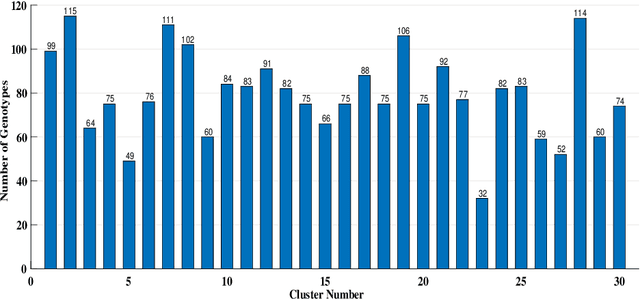
Clustering genotypes based upon their phenotypic characteristics is used to obtain diverse sets of parents that are useful in their breeding programs. The Hierarchical Clustering (HC) algorithm is the current standard in clustering of phenotypic data. This algorithm suffers from low accuracy and high computational complexity issues. To address the accuracy challenge, we propose the use of Spectral Clustering (SC) algorithm. To make the algorithm computationally cheap, we propose using sampling, specifically, Pivotal Sampling that is probability based. Since application of samplings to phenotypic data has not been explored much, for effective comparison, another sampling technique called Vector Quantization (VQ) is adapted for this data as well. VQ has recently given promising results for genome data. The novelty of our SC with Pivotal Sampling algorithm is in constructing the crucial similarity matrix for the clustering algorithm and defining probabilities for the sampling technique. Although our algorithm can be applied to any plant genotypes, we test it on the phenotypic data obtained from about 2400 Soybean genotypes. SC with Pivotal Sampling achieves substantially more accuracy (in terms of Silhouette Values) than all the other proposed competitive clustering with sampling algorithms (i.e. SC with VQ, HC with Pivotal Sampling, and HC with VQ). The complexities of our SC with Pivotal Sampling algorithm and these three variants are almost same because of the involved sampling. In addition to this, SC with Pivotal Sampling outperforms the standard HC algorithm in both accuracy and computational complexity. We experimentally show that we are up to 45% more accurate than HC in terms of clustering accuracy. The computational complexity of our algorithm is more than a magnitude lesser than HC.
Vector Quantized Spectral Clustering applied to Soybean Whole Genome Sequences
Sep 30, 2018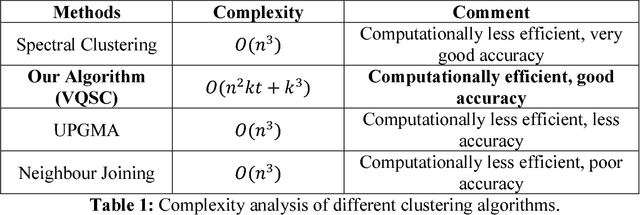
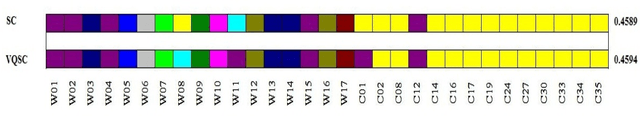
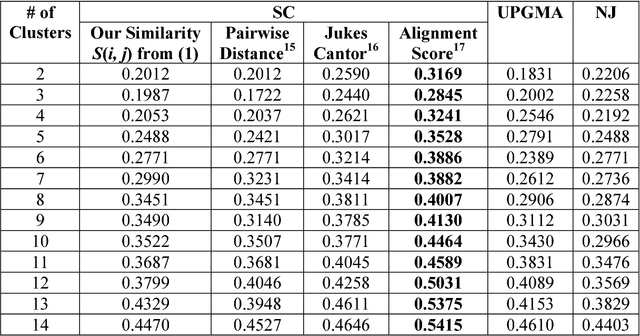
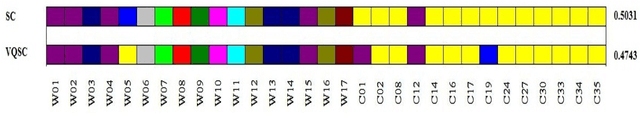
We develop a Vector Quantized Spectral Clustering (VQSC) algorithm that is a combination of Spectral Clustering (SC) and Vector Quantization (VQ) sampling for grouping Soybean genomes. The inspiration here is to use SC for its accuracy and VQ to make the algorithm computationally cheap (the complexity of SC is cubic in-terms of the input size). Although the combination of SC and VQ is not new, the novelty of our work is in developing the crucial similarity matrix in SC as well as use of k-medoids in VQ, both adapted for the Soybean genome data. We compare our approach with commonly used techniques like UPGMA (Un-weighted Pair Graph Method with Arithmetic Mean) and NJ (Neighbour Joining). Experimental results show that our approach outperforms both these techniques significantly in terms of cluster quality (up to 25% better cluster quality) and time complexity (order of magnitude faster).
Localized Multiple Kernel Learning for Anomaly Detection: One-class Classification
Jul 17, 2018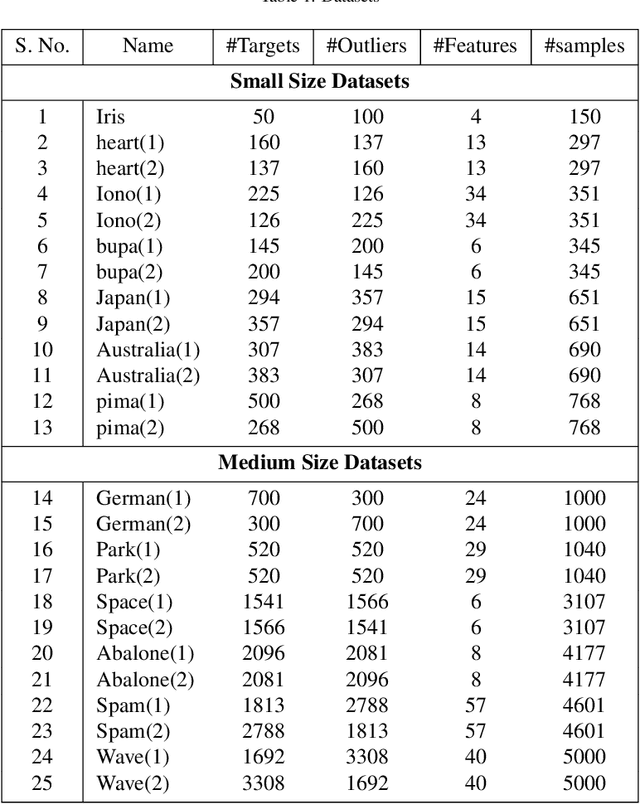
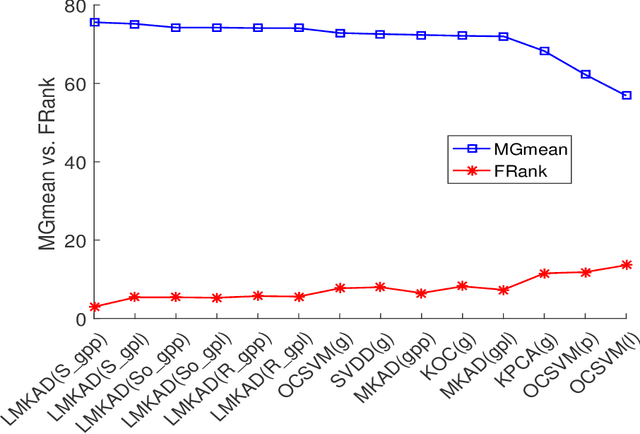
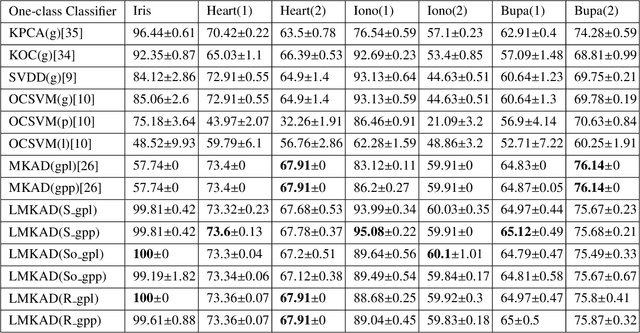
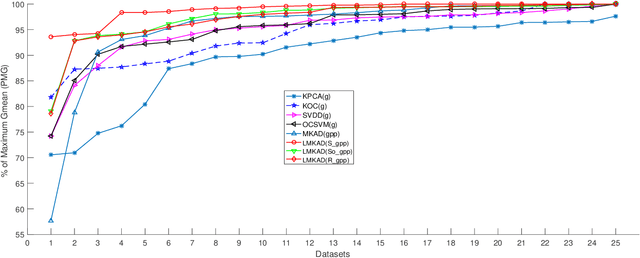
Multi-kernel learning has been well explored in the recent past and has exhibited promising outcomes for multi-class classification and regression tasks. In this paper, we present a multiple kernel learning approach for the One-class Classification (OCC) task and employ it for anomaly detection. Recently, the basic multi-kernel approach has been proposed to solve the OCC problem, which is simply a convex combination of different kernels with equal weights. This paper proposes a Localized Multiple Kernel learning approach for Anomaly Detection (LMKAD) using OCC, where the weight for each kernel is assigned locally. Proposed LMKAD approach adapts the weight for each kernel using a gating function. The parameters of the gating function and one-class classifier are optimized simultaneously through a two-step optimization process. We present the empirical results of the performance of LMKAD on 25 benchmark datasets from various disciplines. This performance is evaluated against existing Multi Kernel Anomaly Detection (MKAD) algorithm, and four other existing kernel-based one-class classifiers to showcase the credibility of our approach. Our algorithm achieves significantly better Gmean scores while using a lesser number of support vectors compared to MKAD. Friedman test is also performed to verify the statistical significance of the results claimed in this paper.