 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Novel Sampled Clustering Algorithm for Rice Phenotypic Data
Dec 22, 2023Phenotypic (or Physical) characteristics of plant species are commonly used to perform clustering. In one of our recent works (Shastri et al. (2021)), we used a probabilistically sampled (using pivotal sampling) and spectrally clustered algorithm to group soybean species. These techniques were used to obtain highly accurate clusterings at a reduced cost. In this work, we extend the earlier algorithm to cluster rice species. We improve the base algorithm in three ways. First, we propose a new function to build the similarity matrix in Spectral Clustering. Commonly, a natural exponential function is used for this purpose. Based upon the spectral graph theory and the involved Cheeger's inequality, we propose the use a base "a" exponential function instead. This gives a similarity matrix spectrum favorable for clustering, which we support via an eigenvalue analysis. Second, the function used to build the similarity matrix in Spectral Clustering was earlier scaled with a fixed factor (called global scaling). Based upon the idea of Zelnik-Manor and Perona (2004), we now use a factor that varies with matrix elements (called local scaling) and works better. Third, to compute the inclusion probability of a specie in the pivotal sampling algorithm, we had earlier used the notion of deviation that captured how far specie's characteristic values were from their respective base values (computed over all species). A maximum function was used before to find the base values. We now use a median function, which is more intuitive. We support this choice using a statistical analysis. With experiments on 1865 rice species, we demonstrate that in terms of silhouette values, our new Sampled Spectral Clustering is 61% better than Hierarchical Clustering (currently prevalent). Also, our new algorithm is significantly faster than Hierarchical Clustering due to the involved sampling.
Probabilistically Sampled and Spectrally Clustered Plant Genotypes using Phenotypic Characteristics
Sep 18, 2020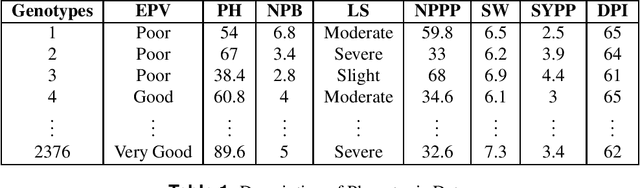
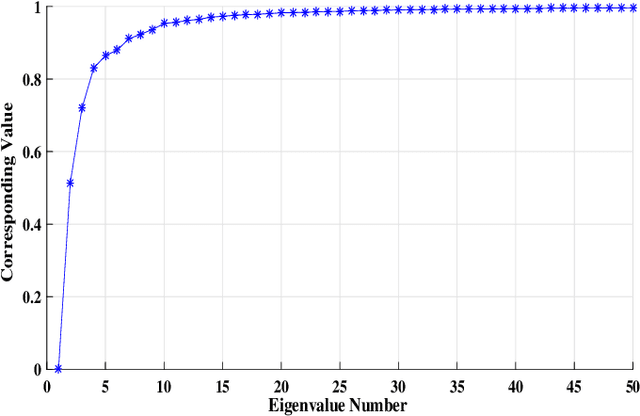
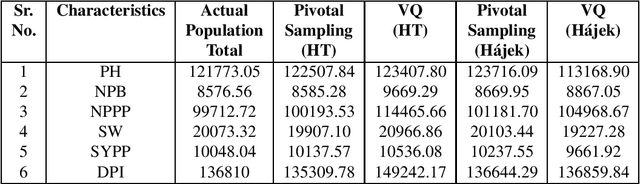
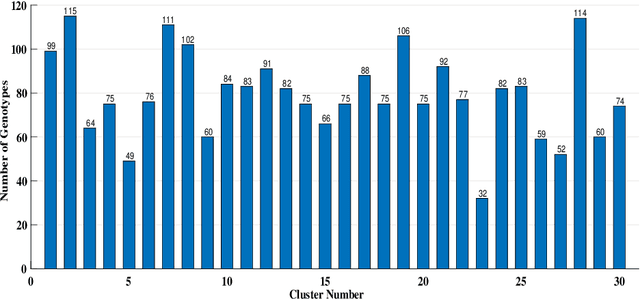
Clustering genotypes based upon their phenotypic characteristics is used to obtain diverse sets of parents that are useful in their breeding programs. The Hierarchical Clustering (HC) algorithm is the current standard in clustering of phenotypic data. This algorithm suffers from low accuracy and high computational complexity issues. To address the accuracy challenge, we propose the use of Spectral Clustering (SC) algorithm. To make the algorithm computationally cheap, we propose using sampling, specifically, Pivotal Sampling that is probability based. Since application of samplings to phenotypic data has not been explored much, for effective comparison, another sampling technique called Vector Quantization (VQ) is adapted for this data as well. VQ has recently given promising results for genome data. The novelty of our SC with Pivotal Sampling algorithm is in constructing the crucial similarity matrix for the clustering algorithm and defining probabilities for the sampling technique. Although our algorithm can be applied to any plant genotypes, we test it on the phenotypic data obtained from about 2400 Soybean genotypes. SC with Pivotal Sampling achieves substantially more accuracy (in terms of Silhouette Values) than all the other proposed competitive clustering with sampling algorithms (i.e. SC with VQ, HC with Pivotal Sampling, and HC with VQ). The complexities of our SC with Pivotal Sampling algorithm and these three variants are almost same because of the involved sampling. In addition to this, SC with Pivotal Sampling outperforms the standard HC algorithm in both accuracy and computational complexity. We experimentally show that we are up to 45% more accurate than HC in terms of clustering accuracy. The computational complexity of our algorithm is more than a magnitude lesser than HC.
Vector Quantized Spectral Clustering applied to Soybean Whole Genome Sequences
Sep 30, 2018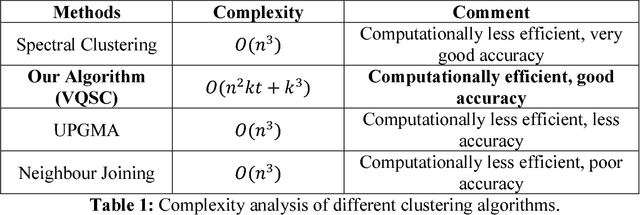
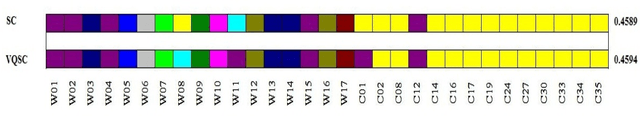
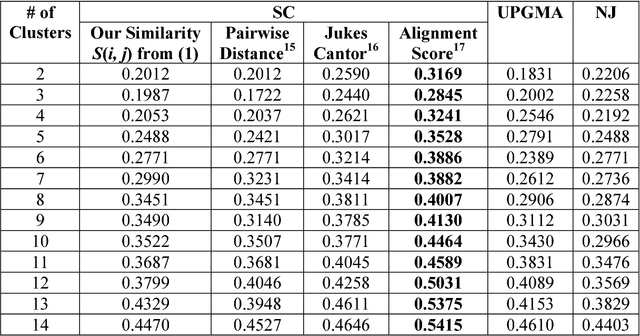
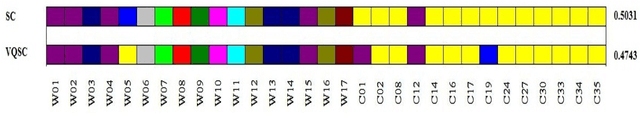
We develop a Vector Quantized Spectral Clustering (VQSC) algorithm that is a combination of Spectral Clustering (SC) and Vector Quantization (VQ) sampling for grouping Soybean genomes. The inspiration here is to use SC for its accuracy and VQ to make the algorithm computationally cheap (the complexity of SC is cubic in-terms of the input size). Although the combination of SC and VQ is not new, the novelty of our work is in developing the crucial similarity matrix in SC as well as use of k-medoids in VQ, both adapted for the Soybean genome data. We compare our approach with commonly used techniques like UPGMA (Un-weighted Pair Graph Method with Arithmetic Mean) and NJ (Neighbour Joining). Experimental results show that our approach outperforms both these techniques significantly in terms of cluster quality (up to 25% better cluster quality) and time complexity (order of magnitude faster).