 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTissue Classification and Whole-Slide Images Analysis via Modeling of the Tumor Microenvironment and Biological Pathways
Jan 13, 2026Automatic integration of whole slide images (WSIs) and gene expression profiles has demonstrated substantial potential in precision clinical diagnosis and cancer progression studies. However, most existing studies focus on individual gene sequences and slide level classification tasks, with limited attention to spatial transcriptomics and patch level applications. To address this limitation, we propose a multimodal network, BioMorphNet, which automatically integrates tissue morphological features and spatial gene expression to support tissue classification and differential gene analysis. For considering morphological features, BioMorphNet constructs a graph to model the relationships between target patches and their neighbors, and adjusts the response strength based on morphological and molecular level similarity, to better characterize the tumor microenvironment. In terms of multimodal interactions, BioMorphNet derives clinical pathway features from spatial transcriptomic data based on a predefined pathway database, serving as a bridge between tissue morphology and gene expression. In addition, a novel learnable pathway module is designed to automatically simulate the biological pathway formation process, providing a complementary representation to existing clinical pathways. Compared with the latest morphology gene multimodal methods, BioMorphNet's average classification metrics improve by 2.67%, 5.48%, and 6.29% for prostate cancer, colorectal cancer, and breast cancer datasets, respectively. BioMorphNet not only classifies tissue categories within WSIs accurately to support tumor localization, but also analyzes differential gene expression between tissue categories based on prediction confidence, contributing to the discovery of potential tumor biomarkers.
Let Samples Speak: Mitigating Spurious Correlation by Exploiting the Clusterness of Samples
Dec 28, 2025Deep learning models are known to often learn features that spuriously correlate with the class label during training but are irrelevant to the prediction task. Existing methods typically address this issue by annotating potential spurious attributes, or filtering spurious features based on some empirical assumptions (e.g., simplicity of bias). However, these methods may yield unsatisfactory performance due to the intricate and elusive nature of spurious correlations in real-world data. In this paper, we propose a data-oriented approach to mitigate the spurious correlation in deep learning models. We observe that samples that are influenced by spurious features tend to exhibit a dispersed distribution in the learned feature space. This allows us to identify the presence of spurious features. Subsequently, we obtain a bias-invariant representation by neutralizing the spurious features based on a simple grouping strategy. Then, we learn a feature transformation to eliminate the spurious features by aligning with this bias-invariant representation. Finally, we update the classifier by incorporating the learned feature transformation and obtain an unbiased model. By integrating the aforementioned identifying, neutralizing, eliminating and updating procedures, we build an effective pipeline for mitigating spurious correlation. Experiments on image and NLP debiasing benchmarks show an improvement in worst group accuracy of more than 20% compared to standard empirical risk minimization (ERM). Codes and checkpoints are available at https://github.com/davelee-uestc/nsf_debiasing .
Spatial Transcriptomics Expression Prediction from Histopathology Based on Cross-Modal Mask Reconstruction and Contrastive Learning
Jun 10, 2025Spatial transcriptomics is a technology that captures gene expression levels at different spatial locations, widely used in tumor microenvironment analysis and molecular profiling of histopathology, providing valuable insights into resolving gene expression and clinical diagnosis of cancer. Due to the high cost of data acquisition, large-scale spatial transcriptomics data remain challenging to obtain. In this study, we develop a contrastive learning-based deep learning method to predict spatially resolved gene expression from whole-slide images. Evaluation across six different disease datasets demonstrates that, compared to existing studies, our method improves Pearson Correlation Coefficient (PCC) in the prediction of highly expressed genes, highly variable genes, and marker genes by 6.27%, 6.11%, and 11.26% respectively. Further analysis indicates that our method preserves gene-gene correlations and applies to datasets with limited samples. Additionally, our method exhibits potential in cancer tissue localization based on biomarker expression.
New Dataset and Methods for Fine-Grained Compositional Referring Expression Comprehension via Specialist-MLLM Collaboration
Feb 28, 2025
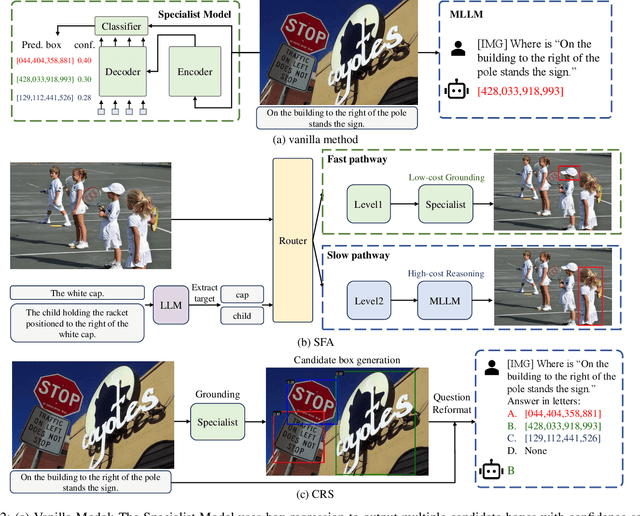


Referring Expression Comprehension (REC) is a foundational cross-modal task that evaluates the interplay of language understanding, image comprehension, and language-to-image grounding. To advance this field, we introduce a new REC dataset with two key features. First, it is designed with controllable difficulty levels, requiring fine-grained reasoning across object categories, attributes, and relationships. Second, it incorporates negative text and images generated through fine-grained editing, explicitly testing a model's ability to reject non-existent targets, an often-overlooked yet critical challenge in existing datasets. To address fine-grained compositional REC, we propose novel methods based on a Specialist-MLLM collaboration framework, leveraging the complementary strengths of them: Specialist Models handle simpler tasks efficiently, while MLLMs are better suited for complex reasoning. Based on this synergy, we introduce two collaborative strategies. The first, Slow-Fast Adaptation (SFA), employs a routing mechanism to adaptively delegate simple tasks to Specialist Models and complex tasks to MLLMs. Additionally, common error patterns in both models are mitigated through a target-refocus strategy. The second, Candidate Region Selection (CRS), generates multiple bounding box candidates based on Specialist Model and uses the advanced reasoning capabilities of MLLMs to identify the correct target. Extensive experiments on our dataset and other challenging compositional benchmarks validate the effectiveness of our approaches. The SFA strategy achieves a trade-off between localization accuracy and efficiency, and the CRS strategy greatly boosts the performance of both Specialist Models and MLLMs. We aim for this work to offer valuable insights into solving complex real-world tasks by strategically combining existing tools for maximum effectiveness, rather than reinventing them.
Multi-level feature fusion network combining attention mechanisms for polyp segmentation
Sep 24, 2023



Clinically, automated polyp segmentation techniques have the potential to significantly improve the efficiency and accuracy of medical diagnosis, thereby reducing the risk of colorectal cancer in patients. Unfortunately, existing methods suffer from two significant weaknesses that can impact the accuracy of segmentation. Firstly, features extracted by encoders are not adequately filtered and utilized. Secondly, semantic conflicts and information redundancy caused by feature fusion are not attended to. To overcome these limitations, we propose a novel approach for polyp segmentation, named MLFF-Net, which leverages multi-level feature fusion and attention mechanisms. Specifically, MLFF-Net comprises three modules: Multi-scale Attention Module (MAM), High-level Feature Enhancement Module (HFEM), and Global Attention Module (GAM). Among these, MAM is used to extract multi-scale information and polyp details from the shallow output of the encoder. In HFEM, the deep features of the encoders complement each other by aggregation. Meanwhile, the attention mechanism redistributes the weight of the aggregated features, weakening the conflicting redundant parts and highlighting the information useful to the task. GAM combines features from the encoder and decoder features, as well as computes global dependencies to prevent receptive field locality. Experimental results on five public datasets show that the proposed method not only can segment multiple types of polyps but also has advantages over current state-of-the-art methods in both accuracy and generalization ability.