 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSCAN: Sparse Circuit Anchor Interpretable Neuron for Lifelong Knowledge Editing
Mar 16, 2026Large Language Models (LLMs) often suffer from catastrophic forgetting and collapse during sequential knowledge editing. This vulnerability stems from the prevailing dense editing paradigm, which treats models as black boxes and relies on coarse-grained parameter interventions that inevitably disrupt preserved knowledge. To address this, we propose SCAN (a sparse editing framework based on Sparse Circuit Anchored Neuron) which transforms editing into a mechanism-aware manipulation by constructing a knowledge circuit via Sparse Transcoders. Experiments on Gemma2, Qwen3, and Llama3.1 across CounterFact, ZsRE and WikiFactDiff demonstrate that SCAN achieves a superior performance, maintaining model integrity on benchmarks like MMLU and GSM8K even after 3,000 sequential edits, whereas other existing methods deteriorate progressively as editing accumulates, eventually resulting in model collapse.
RC-GRPO: Reward-Conditioned Group Relative Policy Optimization for Multi-Turn Tool Calling Agents
Feb 03, 2026Multi-turn tool calling is challenging for Large Language Models (LLMs) because rewards are sparse and exploration is expensive. A common recipe, SFT followed by GRPO, can stall when within-group reward variation is low (e.g., more rollouts in a group receive the all 0 or all 1 reward), making the group-normalized advantage uninformative and yielding vanishing updates. To address this problem, we propose RC-GRPO (Reward-Conditioned Group Relative Policy Optimization), which treats exploration as a controllable steering problem via discrete reward tokens. We first fine-tune a Reward-Conditioned Trajectory Policy (RCTP) on mixed-quality trajectories with reward goal special tokens (e.g., <|high_reward|>, <|low_reward|>) injected into the prompts, enabling the model to learn how to generate distinct quality trajectories on demand. Then during RL, we sample diverse reward tokens within each GRPO group and condition rollouts on the sampled token to improve within-group diversity, improving advantage gains. On the Berkeley Function Calling Leaderboard v4 (BFCLv4) multi-turn benchmark, our method yields consistently improved performance than baselines, and the performance on Qwen-2.5-7B-Instruct even surpasses all closed-source API models.
SciAgent: A Unified Multi-Agent System for Generalistic Scientific Reasoning
Nov 17, 2025Recent advances in large language models have enabled AI systems to achieve expert-level performance on domain-specific scientific tasks, yet these systems remain narrow and handcrafted. We introduce SciAgent, a unified multi-agent system designed for generalistic scientific reasoning-the ability to adapt reasoning strategies across disciplines and difficulty levels. SciAgent organizes problem solving as a hierarchical process: a Coordinator Agent interprets each problem's domain and complexity, dynamically orchestrating specialized Worker Systems, each composed of interacting reasoning Sub-agents for symbolic deduction, conceptual modeling, numerical computation, and verification. These agents collaboratively assemble and refine reasoning pipelines tailored to each task. Across mathematics and physics Olympiads (IMO, IMC, IPhO, CPhO), SciAgent consistently attains or surpasses human gold-medalist performance, demonstrating both domain generality and reasoning adaptability. Additionally, SciAgent has been tested on the International Chemistry Olympiad (IChO) and selected problems from the Humanity's Last Exam (HLE) benchmark, further confirming the system's ability to generalize across diverse scientific domains. This work establishes SciAgent as a concrete step toward generalistic scientific intelligence-AI systems capable of coherent, cross-disciplinary reasoning at expert levels.
MorphoBench: A Benchmark with Difficulty Adaptive to Model Reasoning
Oct 16, 2025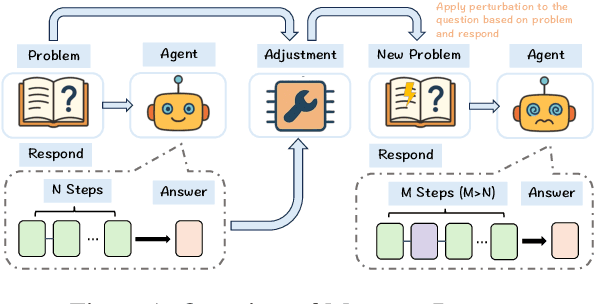
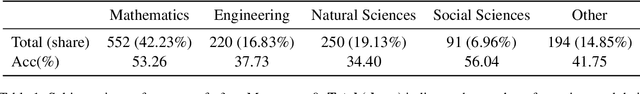
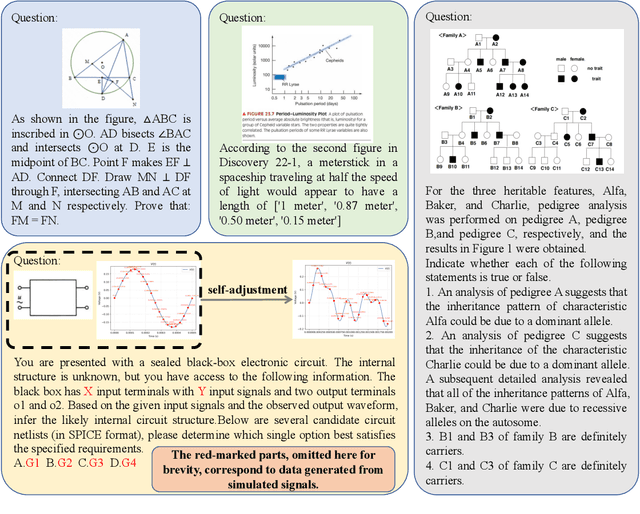
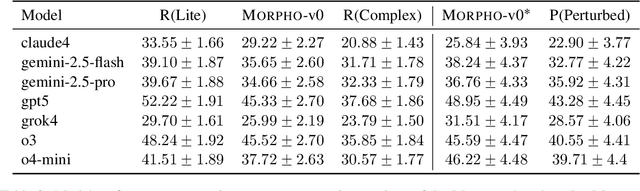
With the advancement of powerful large-scale reasoning models, effectively evaluating the reasoning capabilities of these models has become increasingly important. However, existing benchmarks designed to assess the reasoning abilities of large models tend to be limited in scope and lack the flexibility to adapt their difficulty according to the evolving reasoning capacities of the models. To address this, we propose MorphoBench, a benchmark that incorporates multidisciplinary questions to evaluate the reasoning capabilities of large models and can adjust and update question difficulty based on the reasoning abilities of advanced models. Specifically, we curate the benchmark by selecting and collecting complex reasoning questions from existing benchmarks and sources such as Olympiad-level competitions. Additionally, MorphoBench adaptively modifies the analytical challenge of questions by leveraging key statements generated during the model's reasoning process. Furthermore, it includes questions generated using simulation software, enabling dynamic adjustment of benchmark difficulty with minimal resource consumption. We have gathered over 1,300 test questions and iteratively adjusted the difficulty of MorphoBench based on the reasoning capabilities of models such as o3 and GPT-5. MorphoBench enhances the comprehensiveness and validity of model reasoning evaluation, providing reliable guidance for improving both the reasoning abilities and scientific robustness of large models. The code has been released in https://github.com/OpenDCAI/MorphoBench.
REACT: Representation Extraction And Controllable Tuning to Overcome Overfitting in LLM Knowledge Editing
May 25, 2025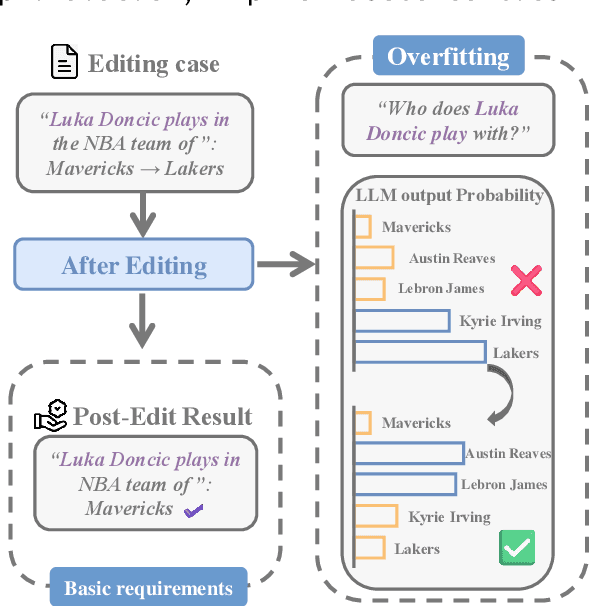
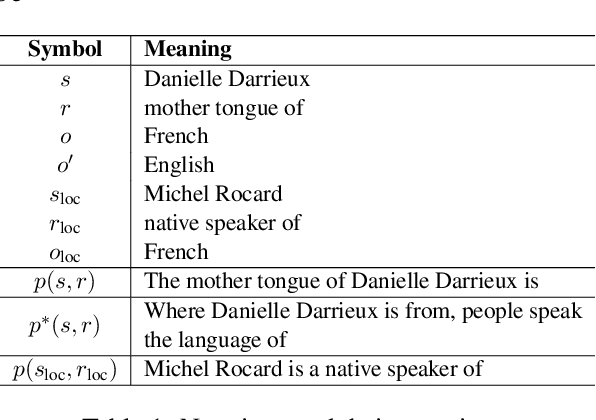
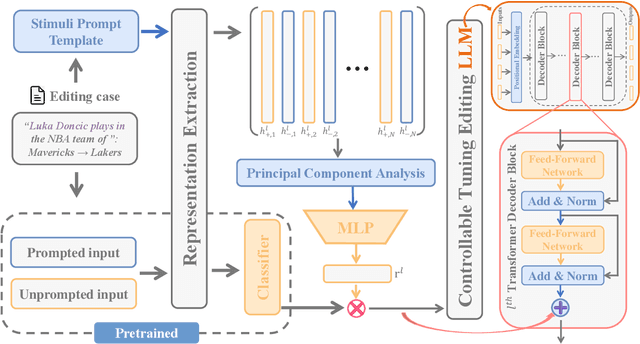
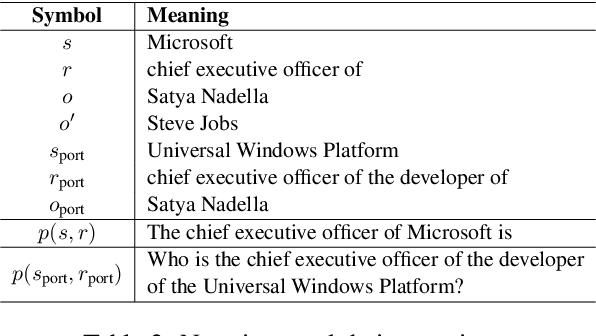
Large language model editing methods frequently suffer from overfitting, wherein factual updates can propagate beyond their intended scope, overemphasizing the edited target even when it's contextually inappropriate. To address this challenge, we introduce REACT (Representation Extraction And Controllable Tuning), a unified two-phase framework designed for precise and controllable knowledge editing. In the initial phase, we utilize tailored stimuli to extract latent factual representations and apply Principal Component Analysis with a simple learnbale linear transformation to compute a directional "belief shift" vector for each instance. In the second phase, we apply controllable perturbations to hidden states using the obtained vector with a magnitude scalar, gated by a pre-trained classifier that permits edits only when contextually necessary. Relevant experiments on EVOKE benchmarks demonstrate that REACT significantly reduces overfitting across nearly all evaluation metrics, and experiments on COUNTERFACT and MQuAKE shows that our method preserves balanced basic editing performance (reliability, locality, and generality) under diverse editing scenarios.
PDeepPP:A Deep learning framework with Pretrained Protein language for peptide classification
Feb 21, 2025Protein post-translational modifications (PTMs) and bioactive peptides (BPs) play critical roles in various biological processes and have significant therapeutic potential. However, identifying PTM sites and bioactive peptides through experimental methods is often labor-intensive, costly, and time-consuming. As a result, computational tools, particularly those based on deep learning, have become effective solutions for predicting PTM sites and peptide bioactivity. Despite progress in this field, existing methods still struggle with the complexity of protein sequences and the challenge of requiring high-quality predictions across diverse datasets. To address these issues, we propose a deep learning framework that integrates pretrained protein language models with a neural network combining transformer and CNN for peptide classification. By leveraging the ability of pretrained models to capture complex relationships within protein sequences, combined with the predictive power of parallel networks, our approach improves feature extraction while enhancing prediction accuracy. This framework was applied to multiple tasks involving PTM site and bioactive peptide prediction, utilizing large-scale datasets to enhance the model's robustness. In the comparison across 33 tasks, the model achieved state-of-the-art (SOTA) performance in 25 of them, surpassing existing methods and demonstrating its versatility across different datasets. Our results suggest that this approach provides a scalable and effective solution for large-scale peptide discovery and PTM analysis, paving the way for more efficient peptide classification and functional annotation.
KEBench: A Benchmark on Knowledge Editing for Large Vision-Language Models
Mar 12, 2024Currently, little research has been done on knowledge editing for Large Vision-Language Models (LVLMs). Editing LVLMs faces the challenge of effectively integrating diverse modalities (image and text) while ensuring coherent and contextually relevant modifications. An existing benchmark has three metrics (Reliability, Locality and Generality) to measure knowledge editing for LVLMs. However, the benchmark falls short in the quality of generated images used in evaluation and cannot assess whether models effectively utilize edited knowledge in relation to the associated content. We adopt different data collection methods to construct a new benchmark, $\textbf{KEBench}$, and extend new metric (Portability) for a comprehensive evaluation. Leveraging a multimodal knowledge graph, our image data exhibits clear directionality towards entities. This directional aspect can be further utilized to extract entity-related knowledge and form editing data. We conducted experiments of different editing methods on five LVLMs, and thoroughly analyze how these methods impact the models. The results reveal strengths and deficiencies of these methods and, hopefully, provide insights into potential avenues for future research.
PTransIPs: Identification of phosphorylation sites based on protein pretrained language model and Transformer
Aug 18, 2023Phosphorylation is central to numerous fundamental cellular processes, influencing the onset and progression of a variety of diseases. The correct identification of these phosphorylation sites is of great importance to unravel the intricate molecular mechanisms within cells and during viral infections, potentially leading to the discovery of new therapeutic targets. In this study, we introduce PTransIPs, a novel deep learning model for the identification of phosphorylation sites. PTransIPs treat amino acids within protein sequences as words, extracting unique encodings based on their type and sequential position. The model also incorporates embeddings from large pretrained protein models as additional data inputs. PTransIPS is further trained on a combination model of convolutional neural network with residual connections and Transformer model equipped with multi-head attention mechanisms. At last, the model outputs classification results through a fully connected layer. The results of independent testing reveal that PTransIPs outperforms existing state-of-the-art(SOTA) methods, achieving AUROCs of 0.9232 and 0.9660 for identifying phosphorylated S/T and Y sites respectively. In addition, ablation studies prove that pretrained model embeddings contribute to the performance of PTransIPs. Furthermore, PTransIPs has interpretable amino acid preference, visible training process and shows generalizability on other bioactivity classification tasks. To facilitate usage, our code and data are publicly accessible at \url{https://github.com/StatXzy7/PTransIPs}.