 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeClifford Kolmogorov-Arnold Networks
Feb 05, 2026We introduce Clifford Kolmogorov-Arnold Network (ClKAN), a flexible and efficient architecture for function approximation in arbitrary Clifford algebra spaces. We propose the use of Randomized Quasi Monte Carlo grid generation as a solution to the exponential scaling associated with higher dimensional algebras. Our ClKAN also introduces new batch normalization strategies to deal with variable domain input. ClKAN finds application in scientific discovery and engineering, and is validated in synthetic and physics inspired tasks.
Logical Guidance for the Exact Composition of Diffusion Models
Feb 05, 2026We propose LOGDIFF (Logical Guidance for the Exact Composition of Diffusion Models), a guidance framework for diffusion models that enables principled constrained generation with complex logical expressions at inference time. We study when exact score-based guidance for complex logical formulas can be obtained from guidance signals associated with atomic properties. First, we derive an exact Boolean calculus that provides a sufficient condition for exact logical guidance. Specifically, if a formula admits a circuit representation in which conjunctions combine conditionally independent subformulas and disjunctions combine subformulas that are either conditionally independent or mutually exclusive, exact logical guidance is achievable. In this case, the guidance signal can be computed exactly from atomic scores and posterior probabilities using an efficient recursive algorithm. Moreover, we show that, for commonly encountered classes of distributions, any desired Boolean formula is compilable into such a circuit representation. Second, by combining atomic guidance scores with posterior probability estimates, we introduce a hybrid guidance approach that bridges classifierguidance and classifier-free guidance, applicable to both compositional logical guidance and standard conditional generation. We demonstrate the effectiveness of our framework on multiple image and protein structure generation tasks.
Variational Kolmogorov-Arnold Network
Jul 03, 2025Kolmogorov Arnold Networks (KANs) are an emerging architecture for building machine learning models. KANs are based on the theoretical foundation of the Kolmogorov-Arnold Theorem and its expansions, which provide an exact representation of a multi-variate continuous bounded function as the composition of a limited number of univariate continuous functions. While such theoretical results are powerful, their use as a representation learning alternative to a multi-layer perceptron (MLP) hinges on the ad-hoc choice of the number of bases modeling each of the univariate functions. In this work, we show how to address this problem by adaptively learning a potentially infinite number of bases for each univariate function during training. We therefore model the problem as a variational inference optimization problem. Our proposal, called InfinityKAN, which uses backpropagation, extends the potential applicability of KANs by treating an important hyperparameter as part of the learning process.
Fast, Modular, and Differentiable Framework for Machine Learning-Enhanced Molecular Simulations
Mar 26, 2025We present an end-to-end differentiable molecular simulation framework (DIMOS) for molecular dynamics and Monte Carlo simulations. DIMOS easily integrates machine-learning-based interatomic potentials and implements classical force fields including particle-mesh Ewald electrostatics. Thanks to its modularity, both classical and machine-learning-based approaches can be easily combined into a hybrid description of the system (ML/MM). By supporting key molecular dynamics features such as efficient neighborlists and constraint algorithms for larger time steps, the framework bridges the gap between hand-optimized simulation engines and the flexibility of a PyTorch implementation. The superior performance and the high versatility is probed in different benchmarks and applications, with speed-up factors of up to $170\times$. The advantage of differentiability is demonstrated by an end-to-end optimization of the proposal distribution in a Markov Chain Monte Carlo simulation based on Hamiltonian Monte Carlo. Using these optimized simulation parameters a $3\times$ acceleration is observed in comparison to ad-hoc chosen simulation parameters. The code is available at https://github.com/nec-research/DIMOS.
Geometric Kolmogorov-Arnold Superposition Theorem
Feb 23, 2025
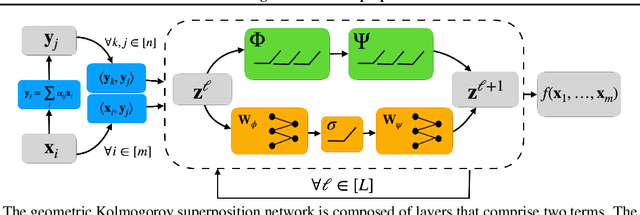
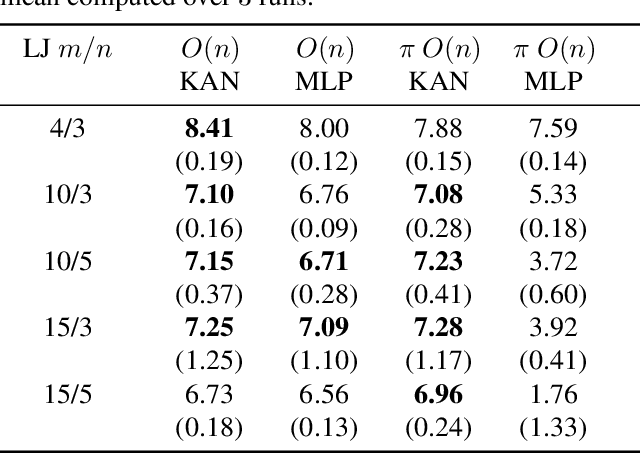
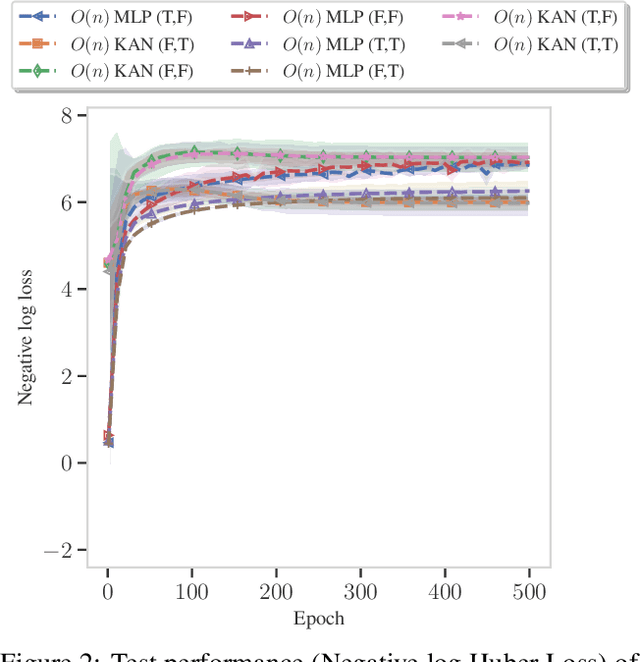
The Kolmogorov-Arnold Theorem (KAT), or more generally, the Kolmogorov Superposition Theorem (KST), establishes that any non-linear multivariate function can be exactly represented as a finite superposition of non-linear univariate functions. Unlike the universal approximation theorem, which provides only an approximate representation without guaranteeing a fixed network size, KST offers a theoretically exact decomposition. The Kolmogorov-Arnold Network (KAN) was introduced as a trainable model to implement KAT, and recent advancements have adapted KAN using concepts from modern neural networks. However, KAN struggles to effectively model physical systems that require inherent equivariance or invariance to $E(3)$ transformations, a key property for many scientific and engineering applications. In this work, we propose a novel extension of KAT and KAN to incorporate equivariance and invariance over $O(n)$ group actions, enabling accurate and efficient modeling of these systems. Our approach provides a unified approach that bridges the gap between mathematical theory and practical architectures for physical systems, expanding the applicability of KAN to a broader class of problems.
Adaptive Width Neural Networks
Jan 27, 2025For almost 70 years, researchers have mostly relied on hyper-parameter tuning to pick the width of neural networks' layers out of many possible choices. This paper challenges the status quo by introducing an easy-to-use technique to learn an unbounded width of a neural network's layer during training. The technique does not rely on alternate optimization nor hand-crafted gradient heuristics; rather, it jointly optimizes the width and the parameters of each layer via simple backpropagation. We apply the technique to a broad range of data domains such as tables, images, texts, and graphs, showing how the width adapts to the task's difficulty. By imposing a soft ordering of importance among neurons, it is possible to truncate the trained network at virtually zero cost, achieving a smooth trade-off between performance and compute resources in a structured way. Alternatively, one can dynamically compress the network with no performance degradation. In light of recent foundation models trained on large datasets, believed to require billions of parameters and where hyper-parameter tuning is unfeasible due to huge training costs, our approach stands as a viable alternative for width learning.
Variational methods for Learning Multilevel Genetic Algorithms using the Kantorovich Monad
Nov 14, 2024



Levels of selection and multilevel evolutionary processes are essential concepts in evolutionary theory, and yet there is a lack of common mathematical models for these core ideas. Here, we propose a unified mathematical framework for formulating and optimizing multilevel evolutionary processes and genetic algorithms over arbitrarily many levels based on concepts from category theory and population genetics. We formulate a multilevel version of the Wright-Fisher process using this approach, and we show that this model can be analyzed to clarify key features of multilevel selection. Particularly, we derive an extended multilevel probabilistic version of Price's Equation via the Kantorovich Monad, and we use this to characterize regimes of parameter space within which selection acts antagonistically or cooperatively across levels. Finally, we show how our framework can provide a unified setting for learning genetic algorithms (GAs), and we show how we can use a Variational Optimization and a multi-level analogue of coalescent analysis to fit multilevel GAs to simulated data.
Graph Reasoning Networks
Jul 08, 2024Graph neural networks (GNNs) are the predominant approach for graph-based machine learning. While neural networks have shown great performance at learning useful representations, they are often criticized for their limited high-level reasoning abilities. In this work, we present Graph Reasoning Networks (GRNs), a novel approach to combine the strengths of fixed and learned graph representations and a reasoning module based on a differentiable satisfiability solver. While results on real-world datasets show comparable performance to GNN, experiments on synthetic datasets demonstrate the potential of the newly proposed method.
Higher-Rank Irreducible Cartesian Tensors for Equivariant Message Passing
May 23, 2024



The ability to perform fast and accurate atomistic simulations is crucial for advancing the chemical sciences. By learning from high-quality data, machine-learned interatomic potentials achieve accuracy on par with ab initio and first-principles methods at a fraction of their computational cost. The success of machine-learned interatomic potentials arises from integrating inductive biases such as equivariance to group actions on an atomic system, e.g., equivariance to rotations and reflections. In particular, the field has notably advanced with the emergence of equivariant message-passing architectures. Most of these models represent an atomic system using spherical tensors, tensor products of which require complicated numerical coefficients and can be computationally demanding. This work introduces higher-rank irreducible Cartesian tensors as an alternative to spherical tensors, addressing the above limitations. We integrate irreducible Cartesian tensor products into message-passing neural networks and prove the equivariance of the resulting layers. Through empirical evaluations on various benchmark data sets, we consistently observe on-par or better performance than that of state-of-the-art spherical models.
Adaptive Message Passing: A General Framework to Mitigate Oversmoothing, Oversquashing, and Underreaching
Dec 27, 2023Long-range interactions are essential for the correct description of complex systems in many scientific fields. The price to pay for including them in the calculations, however, is a dramatic increase in the overall computational costs. Recently, deep graph networks have been employed as efficient, data-driven surrogate models for predicting properties of complex systems represented as graphs. These models rely on a local and iterative message passing strategy that should, in principle, capture long-range information without explicitly modeling the corresponding interactions. In practice, most deep graph networks cannot really model long-range dependencies due to the intrinsic limitations of (synchronous) message passing, namely oversmoothing, oversquashing, and underreaching. This work proposes a general framework that learns to mitigate these limitations: within a variational inference framework, we endow message passing architectures with the ability to freely adapt their depth and filter messages along the way. With theoretical and empirical arguments, we show that this simple strategy better captures long-range interactions, by surpassing the state of the art on five node and graph prediction datasets suited for this problem. Our approach consistently improves the performances of the baselines tested on these tasks. We complement the exposition with qualitative analyses and ablations to get a deeper understanding of the framework's inner workings.