 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeZnTrack -- Data as Code
Jan 19, 2024The past decade has seen tremendous breakthroughs in computation and there is no indication that this will slow any time soon. Machine learning, large-scale computing resources, and increased industry focus have resulted in rising investments in computer-driven solutions for data management, simulations, and model generation. However, with this growth in computation has come an even larger expansion of data and with it, complexity in data storage, sharing, and tracking. In this work, we introduce ZnTrack, a Python-driven data versioning tool. ZnTrack builds upon established version control systems to provide a user-friendly and easy-to-use interface for tracking parameters in experiments, designing workflows, and storing and sharing data. From this ability to reduce large datasets to a simple Python script emerges the concept of Data as Code, a core component of the work presented here and an undoubtedly important concept as the age of computation continues to evolve. ZnTrack offers an open-source, FAIR data compatible Python package to enable users to harness these concepts of the future.
Uncertainty-biased molecular dynamics for learning uniformly accurate interatomic potentials
Dec 03, 2023Efficiently creating a concise but comprehensive data set for training machine-learned interatomic potentials (MLIPs) is an under-explored problem. Active learning (AL), which uses either biased or unbiased molecular dynamics (MD) simulations to generate candidate pools, aims to address this objective. Existing biased and unbiased MD simulations, however, are prone to miss either rare events or extrapolative regions -- areas of the configurational space where unreliable predictions are made. Simultaneously exploring both regions is necessary for developing uniformly accurate MLIPs. In this work, we demonstrate that MD simulations, when biased by the MLIP's energy uncertainty, effectively capture extrapolative regions and rare events without the need to know \textit{a priori} the system's transition temperatures and pressures. Exploiting automatic differentiation, we enhance bias-forces-driven MD simulations by introducing the concept of bias stress. We also employ calibrated ensemble-free uncertainties derived from sketched gradient features to yield MLIPs with similar or better accuracy than ensemble-based uncertainty methods at a lower computational cost. We use the proposed uncertainty-driven AL approach to develop MLIPs for two benchmark systems: alanine dipeptide and MIL-53(Al). Compared to MLIPs trained with conventional MD simulations, MLIPs trained with the proposed data-generation method more accurately represent the relevant configurational space for both atomic systems.
Predicting Properties of Periodic Systems from Cluster Data: A Case Study of Liquid Water
Dec 03, 2023The accuracy of the training data limits the accuracy of bulk properties from machine-learned potentials. For example, hybrid functionals or wave-function-based quantum chemical methods are readily available for cluster data but effectively out-of-scope for periodic structures. We show that local, atom-centred descriptors for machine-learned potentials enable the prediction of bulk properties from cluster model training data, agreeing reasonably well with predictions from bulk training data. We demonstrate such transferability by studying structural and dynamical properties of bulk liquid water with density functional theory and have found an excellent agreement with experimental as well as theoretical counterparts.
Thermally Averaged Magnetic Anisotropy Tensors via Machine Learning Based on Gaussian Moments
Dec 03, 2023



We propose a machine learning method to model molecular tensorial quantities, namely the magnetic anisotropy tensor, based on the Gaussian-moment neural-network approach. We demonstrate that the proposed methodology can achieve an accuracy of 0.3--0.4 cm$^{-1}$ and has excellent generalization capability for out-of-sample configurations. Moreover, in combination with machine-learned interatomic potential energies based on Gaussian moments, our approach can be applied to study the dynamic behavior of magnetic anisotropy tensors and provide a unique insight into spin-phonon relaxation.
Transfer learning for chemically accurate interatomic neural network potentials
Dec 07, 2022



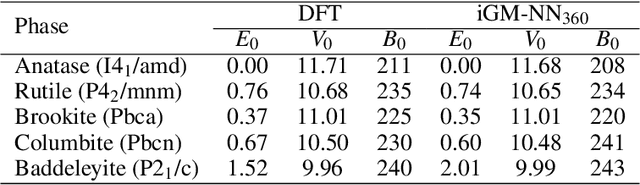
Developing machine learning-based interatomic potentials from ab-initio electronic structure methods remains a challenging task for computational chemistry and materials science. This work studies the capability of transfer learning for efficiently generating chemically accurate interatomic neural network potentials on organic molecules from the MD17 and ANI data sets. We show that pre-training the network parameters on data obtained from density functional calculations considerably improves the sample efficiency of models trained on more accurate ab-initio data. Additionally, we show that fine-tuning with energy labels alone suffices to obtain accurate atomic forces and run large-scale atomistic simulations. We also investigate possible limitations of transfer learning, especially regarding the design and size of the pre-training and fine-tuning data sets. Finally, we provide GM-NN potentials pre-trained and fine-tuned on the ANI-1x and ANI-1ccx data sets, which can easily be fine-tuned on and applied to organic molecules.
A Framework and Benchmark for Deep Batch Active Learning for Regression
Mar 17, 2022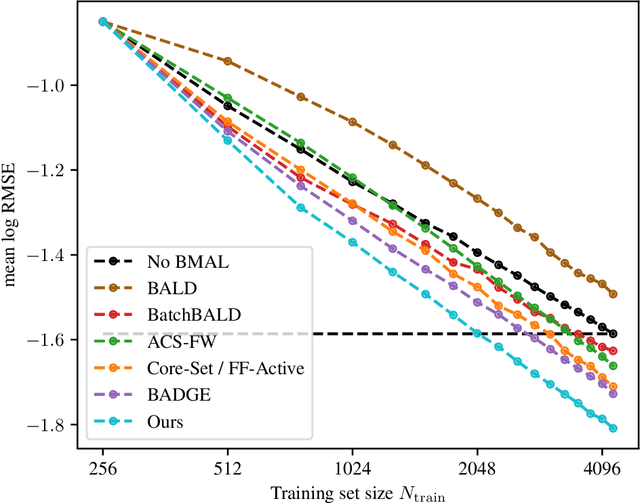


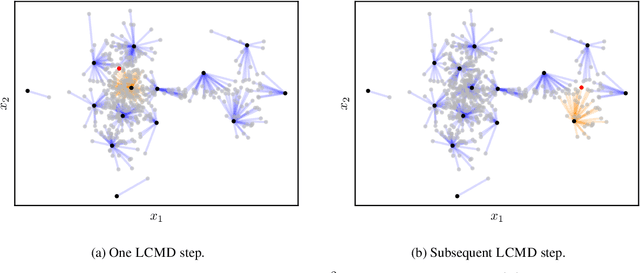
We study the performance of different pool-based Batch Mode Deep Active Learning (BMDAL) methods for regression on tabular data, focusing on methods that do not require to modify the network architecture and training. Our contributions are three-fold: First, we present a framework for constructing BMDAL methods out of kernels, kernel transformations and selection methods, showing that many of the most popular BMDAL methods fit into our framework. Second, we propose new components, leading to a new BMDAL method. Third, we introduce an open-source benchmark with 15 large tabular data sets, which we use to compare different BMDAL methods. Our benchmark results show that a combination of our novel components yields new state-of-the-art results in terms of RMSE and is computationally efficient. We provide open-source code that includes efficient implementations of all kernels, kernel transformations, and selection methods, and can be used for reproducing our results.
Fast and Sample-Efficient Interatomic Neural Network Potentials for Molecules and Materials Based on Gaussian Moments
Sep 20, 2021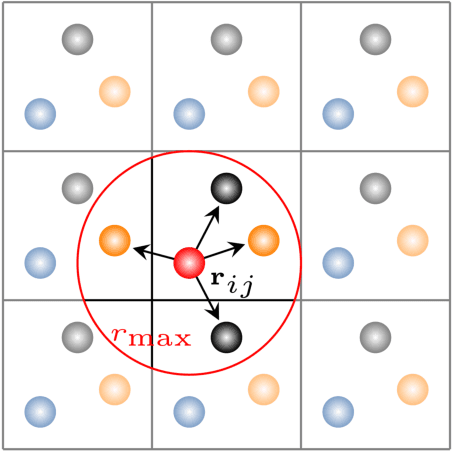
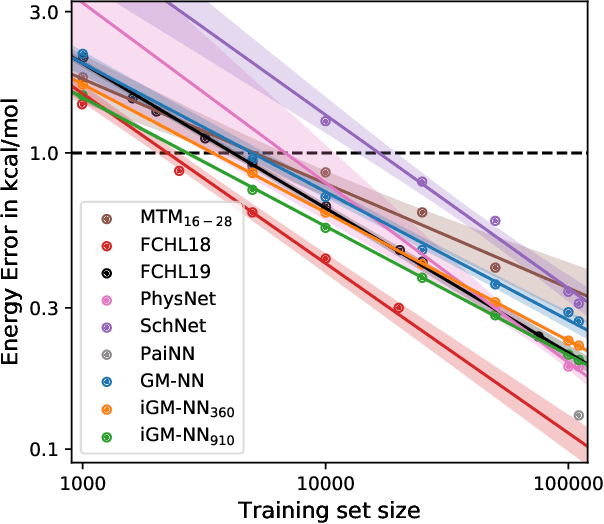
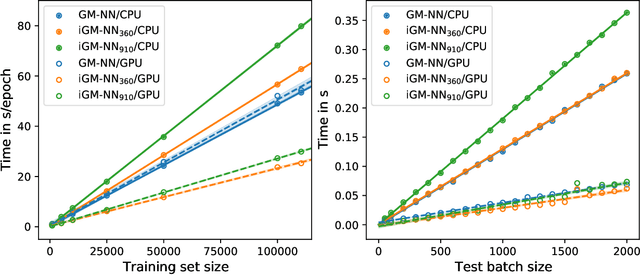
Artificial neural networks (NNs) are one of the most frequently used machine learning approaches to construct interatomic potentials and enable efficient large-scale atomistic simulations with almost ab initio accuracy. However, the simultaneous training of NNs on energies and forces, which are a prerequisite for, e.g., molecular dynamics simulations, can be demanding. In this work, we present an improved NN architecture based on the previous GM-NN model [V. Zaverkin and J. K\"astner, J. Chem. Theory Comput. 16, 5410-5421 (2020)], which shows an improved prediction accuracy and considerably reduced training times. Moreover, we extend the applicability of Gaussian moment-based interatomic potentials to periodic systems and demonstrate the overall excellent transferability and robustness of the respective models. The fast training by the improved methodology is a pre-requisite for training-heavy workflows such as active learning or learning-on-the-fly.
Gaussian Moments as Physically Inspired Molecular Descriptors for Accurate and Scalable Machine Learning Potentials
Sep 15, 2021



Machine learning techniques allow a direct mapping of atomic positions and nuclear charges to the potential energy surface with almost ab-initio accuracy and the computational efficiency of empirical potentials. In this work we propose a machine learning method for constructing high-dimensional potential energy surfaces based on feed-forward neural networks. As input to the neural network we propose an extendable invariant local molecular descriptor constructed from geometric moments. Their formulation via pairwise distance vectors and tensor contractions allows a very efficient implementation on graphical processing units (GPUs). The atomic species is encoded in the molecular descriptor, which allows the restriction to one neural network for the training of all atomic species in the data set. We demonstrate that the accuracy of the developed approach in representing both chemical and configurational spaces is comparable to the one of several established machine learning models. Due to its high accuracy and efficiency, the proposed machine-learned potentials can be used for any further tasks, for example the optimization of molecular geometries, the calculation of rate constants or molecular dynamics.