 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFast and Sample-Efficient Interatomic Neural Network Potentials for Molecules and Materials Based on Gaussian Moments
Paper and Code
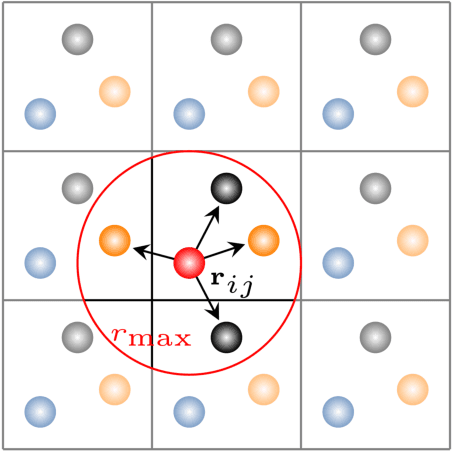
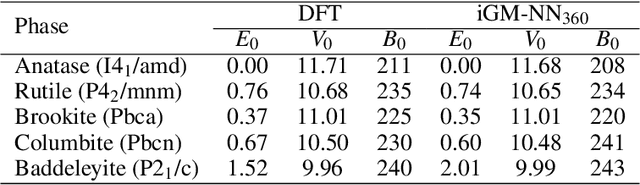
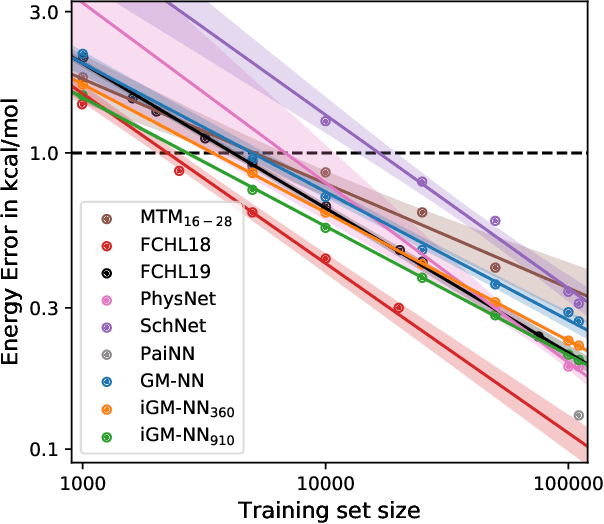
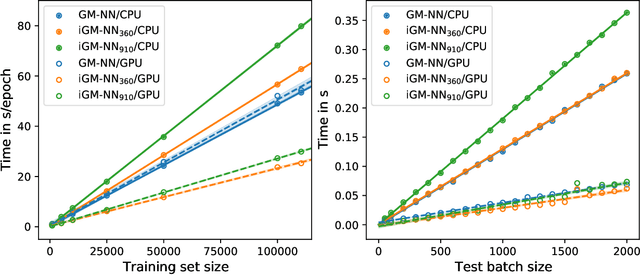
Artificial neural networks (NNs) are one of the most frequently used machine learning approaches to construct interatomic potentials and enable efficient large-scale atomistic simulations with almost ab initio accuracy. However, the simultaneous training of NNs on energies and forces, which are a prerequisite for, e.g., molecular dynamics simulations, can be demanding. In this work, we present an improved NN architecture based on the previous GM-NN model [V. Zaverkin and J. K\"astner, J. Chem. Theory Comput. 16, 5410-5421 (2020)], which shows an improved prediction accuracy and considerably reduced training times. Moreover, we extend the applicability of Gaussian moment-based interatomic potentials to periodic systems and demonstrate the overall excellent transferability and robustness of the respective models. The fast training by the improved methodology is a pre-requisite for training-heavy workflows such as active learning or learning-on-the-fly.