 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHybrid Encoder: Towards Efficient and Precise Native AdsRecommendation via Hybrid Transformer Encoding Networks
Apr 22, 2021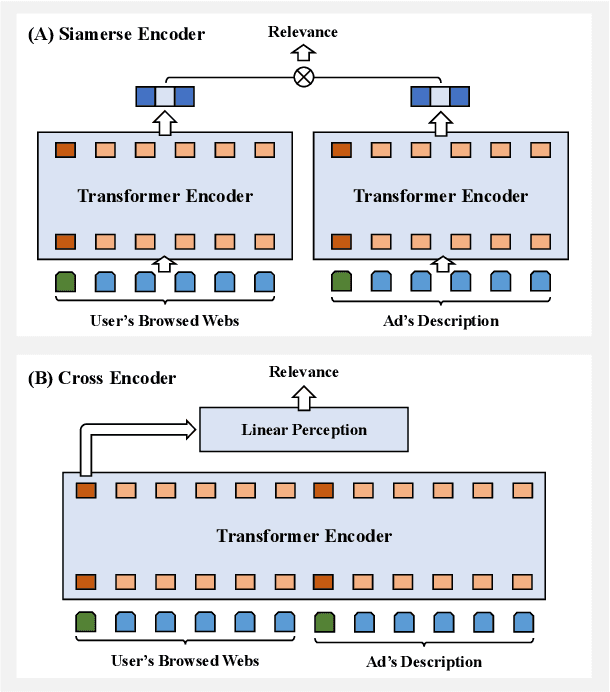
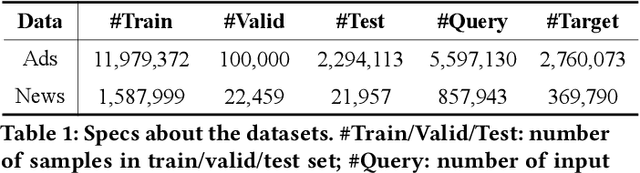
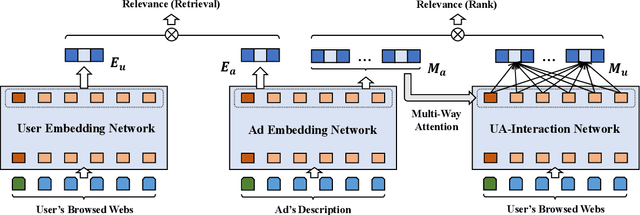
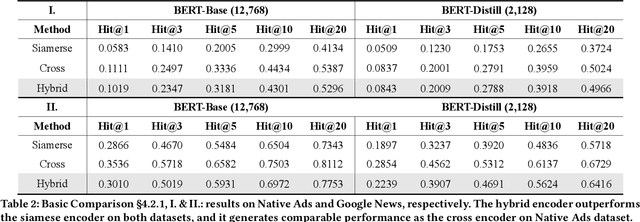
Transformer encoding networks have been proved to be a powerful tool of understanding natural languages. They are playing a critical role in native ads service, which facilitates the recommendation of appropriate ads based on user's web browsing history. For the sake of efficient recommendation, conventional methods would generate user and advertisement embeddings independently with a siamese transformer encoder, such that approximate nearest neighbour search (ANN) can be leveraged. Given that the underlying semantic about user and ad can be complicated, such independently generated embeddings are prone to information loss, which leads to inferior recommendation quality. Although another encoding strategy, the cross encoder, can be much more accurate, it will lead to huge running cost and become infeasible for realtime services, like native ads recommendation. In this work, we propose hybrid encoder, which makes efficient and precise native ads recommendation through two consecutive steps: retrieval and ranking. In the retrieval step, user and ad are encoded with a siamese component, which enables relevant candidates to be retrieved via ANN search. In the ranking step, it further represents each ad with disentangled embeddings and each user with ad-related embeddings, which contributes to the fine-grained selection of high-quality ads from the candidate set. Both steps are light-weighted, thanks to the pre-computed and cached intermedia results. To optimize the hybrid encoder's performance in this two-stage workflow, a progressive training pipeline is developed, which builds up the model's capability in the retrieval and ranking task step-by-step. The hybrid encoder's effectiveness is experimentally verified: with very little additional cost, it outperforms the siamese encoder significantly and achieves comparable recommendation quality as the cross encoder.
Multi-Interest-Aware User Modeling for Large-Scale Sequential Recommendations
Mar 04, 2021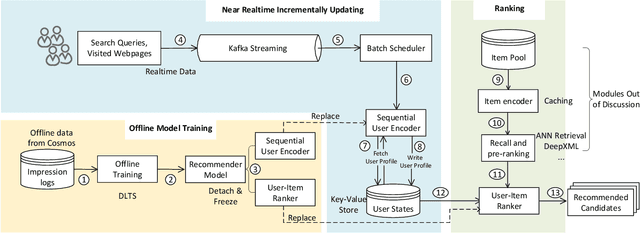
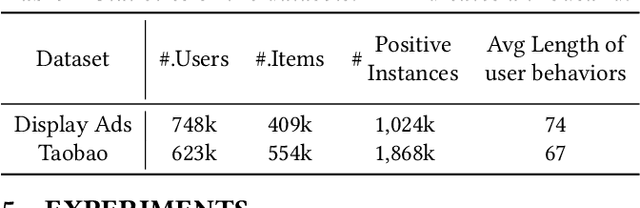
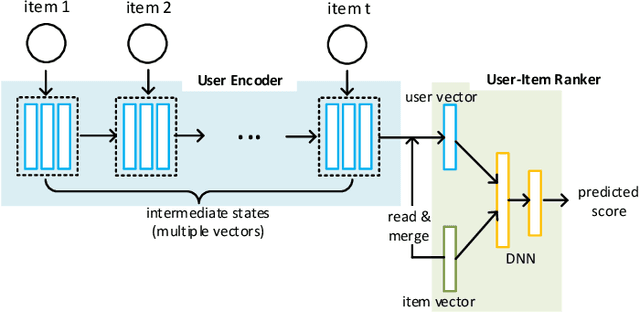
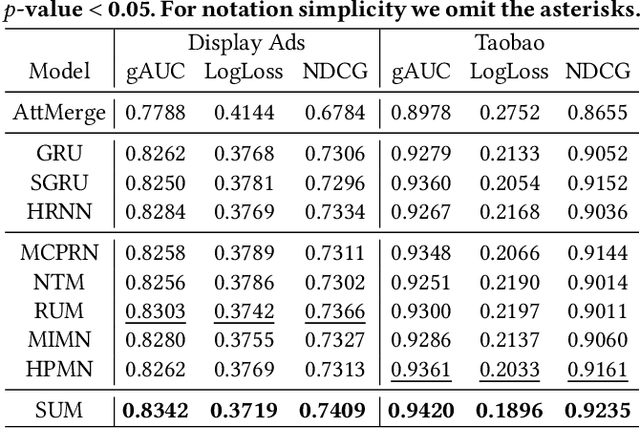
Precise user modeling is critical for online personalized recommendation services. Generally, users' interests are diverse and are not limited to a single aspect, which is particularly evident when their behaviors are observed for a longer time. For example, a user may demonstrate interests in cats/dogs, dancing and food \& delights when browsing short videos on Tik Tok; the same user may show interests in real estate and women's wear in her web browsing behaviors. Traditional models tend to encode a user's behaviors into a single embedding vector, which do not have enough capacity to effectively capture her diverse interests. This paper proposes a Sequential User Matrix (SUM) to accurately and efficiently capture users' diverse interests. SUM models user behavior with a multi-channel network, with each channel representing a different aspect of the user's interests. User states in different channels are updated by an \emph{erase-and-add} paradigm with interest- and instance-level attention. We further propose a local proximity debuff component and a highway connection component to make the model more robust and accurate. SUM can be maintained and updated incrementally, making it feasible to be deployed for large-scale online serving. We conduct extensive experiments on two datasets. Results demonstrate that SUM consistently outperforms state-of-the-art baselines.
A powerful and efficient set test for genetic markers that handles confounders
Apr 08, 2013
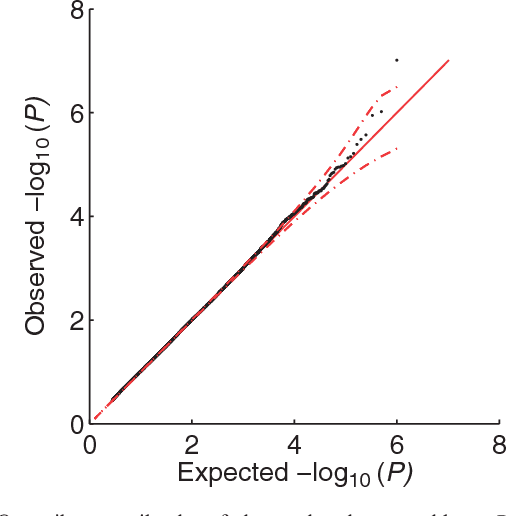
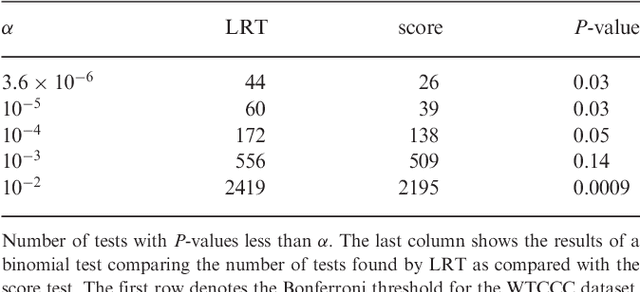

Approaches for testing sets of variants, such as a set of rare or common variants within a gene or pathway, for association with complex traits are important. In particular, set tests allow for aggregation of weak signal within a set, can capture interplay among variants, and reduce the burden of multiple hypothesis testing. Until now, these approaches did not address confounding by family relatedness and population structure, a problem that is becoming more important as larger data sets are used to increase power. Results: We introduce a new approach for set tests that handles confounders. Our model is based on the linear mixed model and uses two random effects-one to capture the set association signal and one to capture confounders. We also introduce a computational speedup for two-random-effects models that makes this approach feasible even for extremely large cohorts. Using this model with both the likelihood ratio test and score test, we find that the former yields more power while controlling type I error. Application of our approach to richly structured GAW14 data demonstrates that our method successfully corrects for population structure and family relatedness, while application of our method to a 15,000 individual Crohn's disease case-control cohort demonstrates that it additionally recovers genes not recoverable by univariate analysis. Availability: A Python-based library implementing our approach is available at http://mscompbio.codeplex.com