 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA powerful and efficient set test for genetic markers that handles confounders
Apr 08, 2013
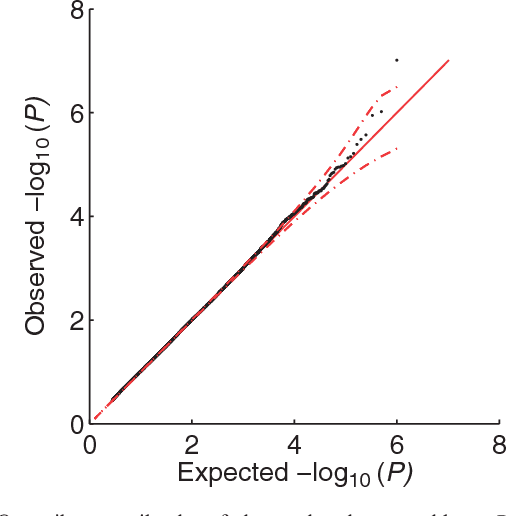
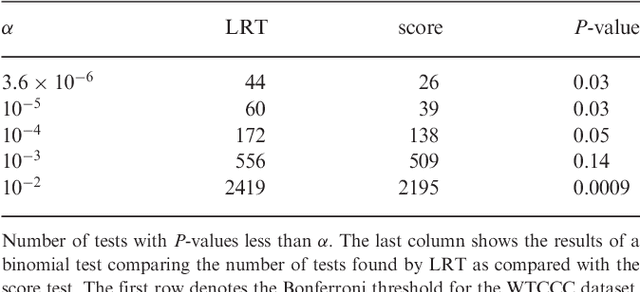

Approaches for testing sets of variants, such as a set of rare or common variants within a gene or pathway, for association with complex traits are important. In particular, set tests allow for aggregation of weak signal within a set, can capture interplay among variants, and reduce the burden of multiple hypothesis testing. Until now, these approaches did not address confounding by family relatedness and population structure, a problem that is becoming more important as larger data sets are used to increase power. Results: We introduce a new approach for set tests that handles confounders. Our model is based on the linear mixed model and uses two random effects-one to capture the set association signal and one to capture confounders. We also introduce a computational speedup for two-random-effects models that makes this approach feasible even for extremely large cohorts. Using this model with both the likelihood ratio test and score test, we find that the former yields more power while controlling type I error. Application of our approach to richly structured GAW14 data demonstrates that our method successfully corrects for population structure and family relatedness, while application of our method to a 15,000 individual Crohn's disease case-control cohort demonstrates that it additionally recovers genes not recoverable by univariate analysis. Availability: A Python-based library implementing our approach is available at http://mscompbio.codeplex.com