 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeJoint registration and synthesis using a probabilistic model for alignment of MRI and histological sections
Jan 16, 2018
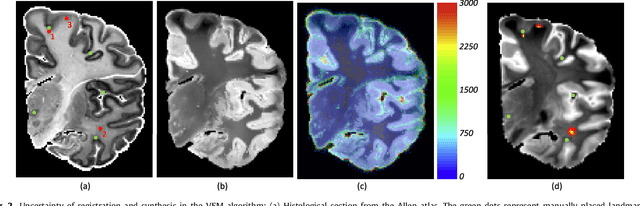
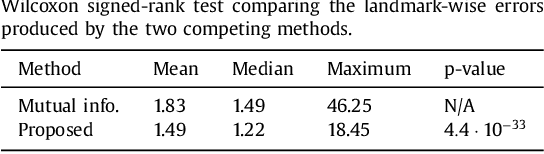
Nonlinear registration of 2D histological sections with corresponding slices of MRI data is a critical step of 3D histology reconstruction. This task is difficult due to the large differences in image contrast and resolution, as well as the complex nonrigid distortions produced when sectioning the sample and mounting it on the glass slide. It has been shown in brain MRI registration that better spatial alignment across modalities can be obtained by synthesizing one modality from the other and then using intra-modality registration metrics, rather than by using mutual information (MI) as metric. However, such an approach typically requires a database of aligned images from the two modalities, which is very difficult to obtain for histology/MRI. Here, we overcome this limitation with a probabilistic method that simultaneously solves for registration and synthesis directly on the target images, without any training data. In our model, the MRI slice is assumed to be a contrast-warped, spatially deformed version of the histological section. We use approximate Bayesian inference to iteratively refine the probabilistic estimate of the synthesis and the registration, while accounting for each other's uncertainty. Moreover, manually placed landmarks can be seamlessly integrated in the framework for increased performance. Experiments on a synthetic dataset show that, compared with MI, the proposed method makes it possible to use a much more flexible deformation model in the registration to improve its accuracy, without compromising robustness. Moreover, our framework also exploits information in manually placed landmarks more efficiently than MI, since landmarks inform both synthesis and registration - as opposed to registration alone. Finally, we show qualitative results on the public Allen atlas, in which the proposed method provides a clear improvement over MI based registration.
Discovering Neuronal Cell Types and Their Gene Expression Profiles Using a Spatial Point Process Mixture Model
Jun 11, 2016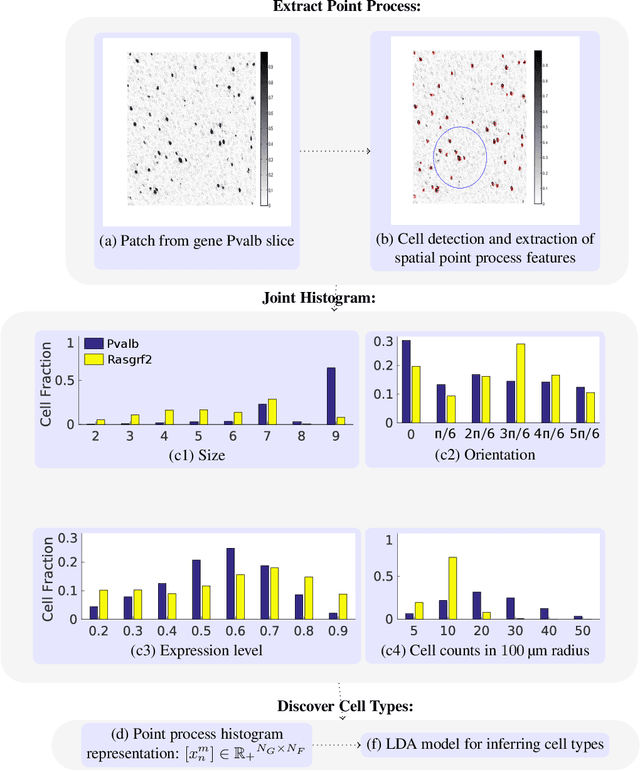
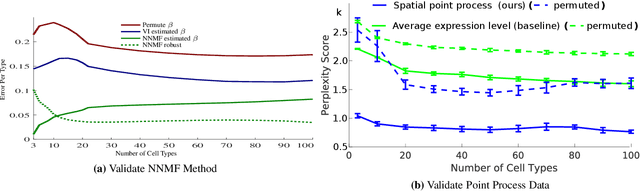
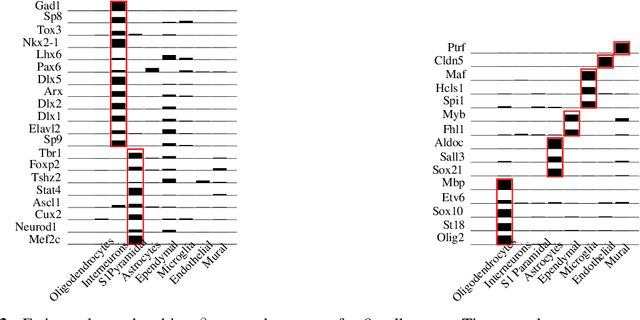
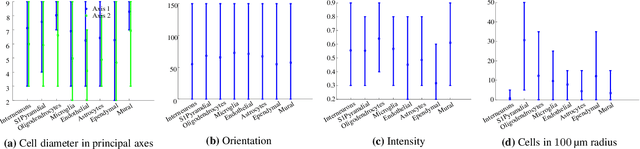
Cataloging the neuronal cell types that comprise circuitry of individual brain regions is a major goal of modern neuroscience and the BRAIN initiative. Single-cell RNA sequencing can now be used to measure the gene expression profiles of individual neurons and to categorize neurons based on their gene expression profiles. While the single-cell techniques are extremely powerful and hold great promise, they are currently still labor intensive, have a high cost per cell, and, most importantly, do not provide information on spatial distribution of cell types in specific regions of the brain. We propose a complementary approach that uses computational methods to infer the cell types and their gene expression profiles through analysis of brain-wide single-cell resolution in situ hybridization (ISH) imagery contained in the Allen Brain Atlas (ABA). We measure the spatial distribution of neurons labeled in the ISH image for each gene and model it as a spatial point process mixture, whose mixture weights are given by the cell types which express that gene. By fitting a point process mixture model jointly to the ISH images, we infer both the spatial point process distribution for each cell type and their gene expression profile. We validate our predictions of cell type-specific gene expression profiles using single cell RNA sequencing data, recently published for the mouse somatosensory cortex. Jointly with the gene expression profiles, cell features such as cell size, orientation, intensity and local density level are inferred per cell type.