 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRepresenting Time Series as Structured Programs for LLM Reasoning
Jun 10, 2026Large language models (LLMs) have demonstrated strong reasoning and instruction-following capabilities, making them potentially powerful tools for time-series analysis. However, time series lie outside their native textual modality, raising a fundamental question: how should time series be represented so that LLMs can reason about them effectively? Existing work typically serializes raw numerical sequences or fine-tunes pre-trained LLMs on time-series data. These approaches place the burden of extracting temporal structure directly on the LLM, creating a modality mismatch that often degrades performance on long sequences and introduces substantial computational overhead. In this work, we introduce Time-Series-to-Structured-Program representation (T2SP), a deterministic, training-free method that represents a time series as a structured symbolic program. T2SP decomposes time series into trends, periods, and salient events, expressing them in a program-friendly format aligned with the textual and code-like modalities on which LLMs are natively trained. By shifting temporal-structure extraction from the model to the representation itself, T2SP enables off-the-shelf LLMs to leverage their existing reasoning capabilities for time-series understanding. We evaluate T2SP on three reasoning tasks -- editing, captioning, and question answering -- where it consistently improves performance, reduces reasoning time, and lowers failure rates compared with raw-string representations. Our results demonstrate that T2SP provides an effective interface between time series and LLMs.
Explaining Black-Box Language Models: Learning to Optimize Linguistically-Structured Word Subsets
Jun 07, 2026As deep language models (DLMs) are increasingly deployed in high-stakes domains such as healthcare, understanding their decision rationale becomes paramount for ensuring trust, safety, and accountability. However, achieving this vital level of interpretability is particularly challenging when these DLMs operate as black-box systems (e.g., via APIs), where access to internal model states (e.g., parameters, gradients) is restricted. Despite numerous efforts, existing explanation methods often fail to concurrently satisfy three key desiderata: (i) inference-time efficiency, (ii) black-box compatibility without inducing out-of-distribution behavior, and (iii) comprehensible explanations grounded in the input's linguistic structure. To address these challenges, we propose a method that explains predictions of DLMs by selecting a small, informative subset of input words. We formulate this as an amortized optimization problem, enabling efficient one-shot inference without the need for input-specific search. Our selection policy is trained via REINFORCE-style policy gradients, allowing discrete word selection in a fully gradient-free setting. To enhance interpretability and align with human linguistic intuition, we integrate graph-structured knowledge into this selection process, fostering linguistically coherent subsets that result in explanations both highly informative and cognitively meaningful to end-users. We evaluated our method on diverse DLM architectures and multiple real-world datasets. It consistently identifies word subsets with enhanced discriminative power and stronger alignment with linguistically salient cues, outperforming both conventional black-box compatible methods and gradient-based approaches that are given oracle access to the black-box model's gradients for a more challenging benchmark. Our code is available at here.
Agentic Molecular Recovery via Molecule-Aware Exploration
Jun 04, 2026Text-guided molecular generation with LLMs often yields invalid SMILES. We argue that invalid drafts should be addressed through a shift from validity-oriented repair to identity-preserving molecular recovery: the objective is not only to restore chemical validity, but also to preserve target-relevant structural cues and recover the molecular identity implied by the description. This perspective reveals the limitations of existing correction strategies. Post-hoc repair can recover validity while distorting key structures, LLM-only correction can introduce unintended global drift, and generic agentic correction remains constrained by greedy single-candidate trajectories even when equipped with executable RDKit edit tools. To address these limitations, we propose AMREC, which couples molecule-aware mismatch tracking with expanded candidate exploration and trajectory-level selection. On invalid ChEBI-20 drafts from three backbone models, AMREC achieves the strongest overall recovery profile across structural, exact-match, and string-level metrics.
Localizing Input Uncertainty Quantification for Large Language Models via Shapley Values
May 27, 2026As large language models (LLMs) are increasingly integrated into high-stakes decision-making, the ability to reliably quantify uncertainty has become a critical requirement for safety and trust. However, current uncertainty quantification methods primarily operate at the output level, often failing to distinguish whether uncertainty arises from the model's lack of knowledge or from ambiguity in the user's input. While input-centric uncertainty quantification has recently emerged as a promising direction, it remains relatively underexplored and typically relies on coarse, input-level information. Consequently, users are provided with scalar uncertainty scores that offer little actionable guidance on which parts of the input should be clarified to improve reliability. To address this limitation, we propose Shapley-based input uncertainty Quantification (ShaQ), a framework for span-level attribution of input-induced uncertainty. Our approach models ambiguous spans in the input as players in a cooperative game and quantifies their contributions using Shapley values, defined via the weighted average of marginal reductions in conditional entropy obtained by clarifying each span coalition. Unlike existing input-level approaches, our formulation captures complex interactions among spans and provides a principled decomposition in which individual attributions sum exactly to the total input-induced uncertainty. We evaluate ShaQ on the AmbigQA and AmbiEnt benchmarks, where it achieves state-of-the-art performance in ambiguity detection. We further demonstrate its utility on MediTOD, showing that ShaQ can localize under-specified clinical utterances and facilitate human-AI collaboration in high-stakes settings. Overall, ShaQ improves uncertainty estimation and provides actionable insights for targeted input clarification.
INSHAPE: Instance-Level Shapelets for Interpretable Time-Series Classification
May 19, 2026Discovering shapelets -- i.e., discriminative temporal patterns within time series -- has been widely studied to address the inherent complexity of time-series classification (TSC) and to make model decision-making processes more transparent. However, existing methods primarily focus on population-level shapelets optimized across the entire dataset, which leads to two fundamental limitations: (i) population-level patterns often misalign with instance-specific features, resulting in suboptimal performance and potentially misleading interpretations, and (ii) most methods treat shapelets as independent entities, overlooking important temporal dependencies and interactions among multiple patterns. To address these limitations, we propose INSHAPE, an interpretable TSC framework that discovers variable-length, discriminative temporal patterns specific to each time series. INSHAPE identifies these patterns as non-overlapping segments and models their temporal dependencies, thereby providing clear instance-level interpretations while achieving strong predictive performance. Furthermore, INSHAPE bridges local and global interpretability through a bottom-up approach, aggregating instance-level shapelets into prototypical (population-level) shapelets. Extensive experiments on 128 UCR and 30 UEA benchmark datasets show that INSHAPE consistently outperforms state-of-the-art shapelet-based methods while providing more intuitive and interpretable insights.
PETAR: Localized Findings Generation with Mask-Aware Vision-Language Modeling for PET Automated Reporting
Oct 31, 2025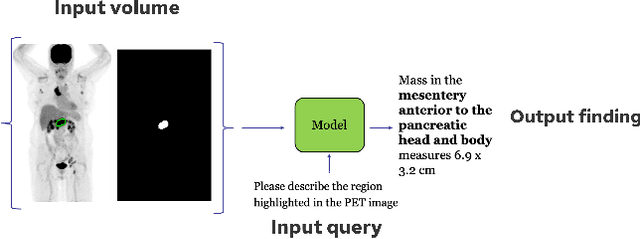


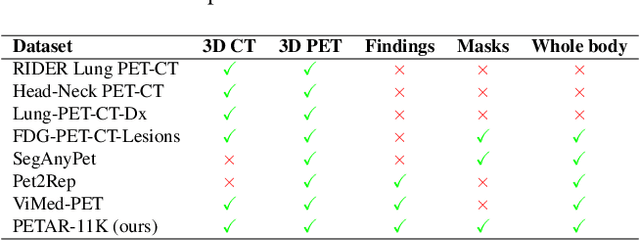
Recent advances in vision-language models (VLMs) have enabled impressive multimodal reasoning, yet most medical applications remain limited to 2D imaging. In this work, we extend VLMs to 3D positron emission tomography and computed tomography (PET/CT), a domain characterized by large volumetric data, small and dispersed lesions, and lengthy radiology reports. We introduce a large-scale dataset comprising over 11,000 lesion-level descriptions paired with 3D segmentations from more than 5,000 PET/CT exams, extracted via a hybrid rule-based and large language model (LLM) pipeline. Building upon this dataset, we propose PETAR-4B, a 3D mask-aware vision-language model that integrates PET, CT, and lesion contours for spatially grounded report generation. PETAR bridges global contextual reasoning with fine-grained lesion awareness, producing clinically coherent and localized findings. Comprehensive automated and human evaluations demonstrate that PETAR substantially improves PET/CT report generation quality, advancing 3D medical vision-language understanding.
BioBridge: Unified Bio-Embedding with Bridging Modality in Code-Switched EMR
Dec 16, 2024
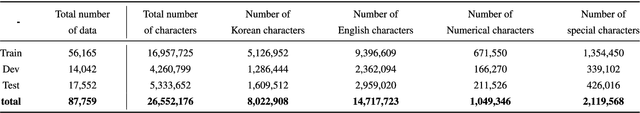
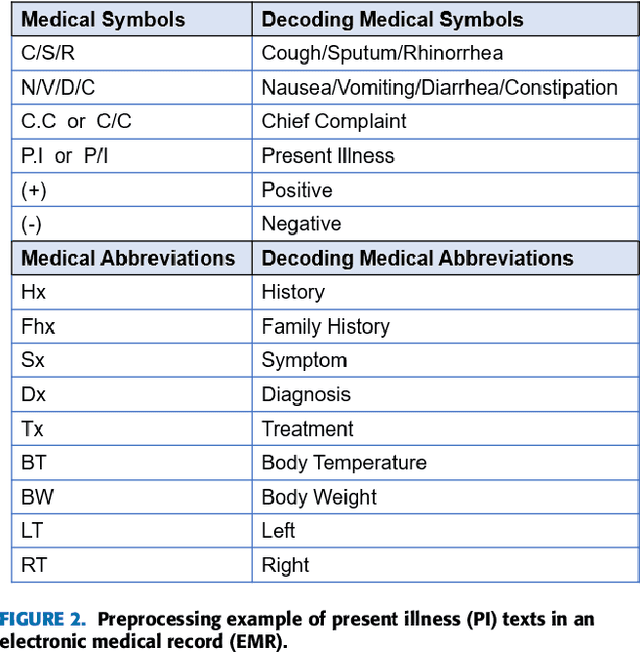
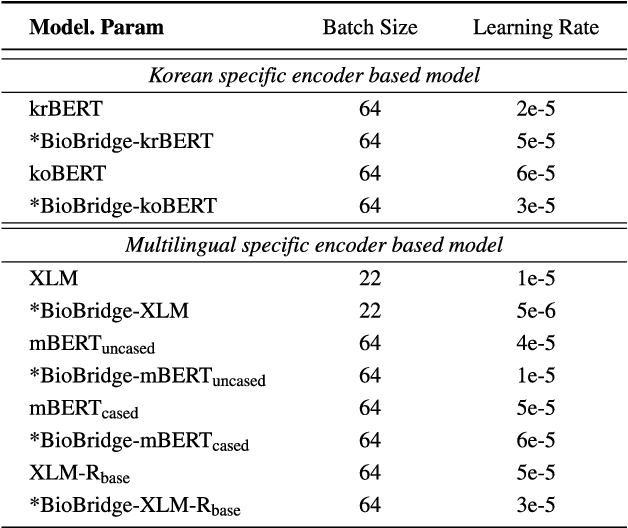
Pediatric Emergency Department (PED) overcrowding presents a significant global challenge, prompting the need for efficient solutions. This paper introduces the BioBridge framework, a novel approach that applies Natural Language Processing (NLP) to Electronic Medical Records (EMRs) in written free-text form to enhance decision-making in PED. In non-English speaking countries, such as South Korea, EMR data is often written in a Code-Switching (CS) format that mixes the native language with English, with most code-switched English words having clinical significance. The BioBridge framework consists of two core modules: "bridging modality in context" and "unified bio-embedding." The "bridging modality in context" module improves the contextual understanding of bilingual and code-switched EMRs. In the "unified bio-embedding" module, the knowledge of the model trained in the medical domain is injected into the encoder-based model to bridge the gap between the medical and general domains. Experimental results demonstrate that the proposed BioBridge significantly performance traditional machine learning and pre-trained encoder-based models on several metrics, including F1 score, area under the receiver operating characteristic curve (AUROC), area under the precision-recall curve (AUPRC), and Brier score. Specifically, BioBridge-XLM achieved enhancements of 0.85% in F1 score, 0.75% in AUROC, and 0.76% in AUPRC, along with a notable 3.04% decrease in the Brier score, demonstrating marked improvements in accuracy, reliability, and prediction calibration over the baseline XLM model. The source code will be made publicly available.
* Accepted at IEEE Access 2024
Toward a Well-Calibrated Discrimination via Survival Outcome-Aware Contrastive Learning
Oct 15, 2024Previous deep learning approaches for survival analysis have primarily relied on ranking losses to improve discrimination performance, which often comes at the expense of calibration performance. To address such an issue, we propose a novel contrastive learning approach specifically designed to enhance discrimination \textit{without} sacrificing calibration. Our method employs weighted sampling within a contrastive learning framework, assigning lower penalties to samples with similar survival outcomes. This aligns well with the assumption that patients with similar event times share similar clinical statuses. Consequently, when augmented with the commonly used negative log-likelihood loss, our approach significantly improves discrimination performance without directly manipulating the model outputs, thereby achieving better calibration. Experiments on multiple real-world clinical datasets demonstrate that our method outperforms state-of-the-art deep survival models in both discrimination and calibration. Through comprehensive ablation studies, we further validate the effectiveness of our approach through quantitative and qualitative analyses.
Enhancing Anomaly Detection via Generating Diversified and Hard-to-distinguish Synthetic Anomalies
Sep 16, 2024Unsupervised anomaly detection is a daunting task, as it relies solely on normality patterns from the training data to identify unseen anomalies during testing. Recent approaches have focused on leveraging domain-specific transformations or perturbations to generate synthetic anomalies from normal samples. The objective here is to acquire insights into normality patterns by learning to differentiate between normal samples and these crafted anomalies. However, these approaches often encounter limitations when domain-specific transformations are not well-specified such as in tabular data, or when it becomes trivial to distinguish between them. To address these issues, we introduce a novel domain-agnostic method that employs a set of conditional perturbators and a discriminator. The perturbators are trained to generate input-dependent perturbations, which are subsequently utilized to construct synthetic anomalies, and the discriminator is trained to distinguish normal samples from them. We ensure that the generated anomalies are both diverse and hard to distinguish through two key strategies: i) directing perturbations to be orthogonal to each other and ii) constraining perturbations to remain in proximity to normal samples. Throughout experiments on real-world datasets, we demonstrate the superiority of our method over state-of-the-art benchmarks, which is evident not only in image data but also in tabular data, where domain-specific transformation is not readily accessible. Additionally, we empirically confirm the adaptability of our method to semi-supervised settings, demonstrating its capacity to incorporate supervised signals to enhance anomaly detection performance even further.
Automatic Quantification of Serial PET/CT Images for Pediatric Hodgkin Lymphoma Patients Using a Longitudinally-Aware Segmentation Network
Apr 12, 2024


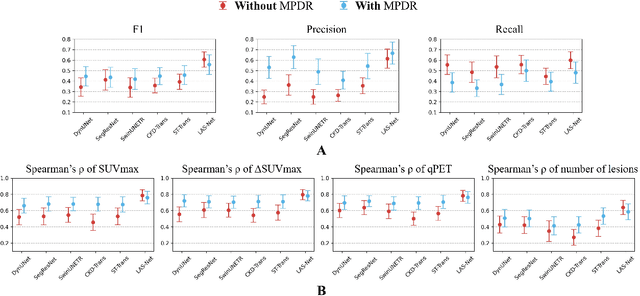
$\textbf{Purpose}$: Automatic quantification of longitudinal changes in PET scans for lymphoma patients has proven challenging, as residual disease in interim-therapy scans is often subtle and difficult to detect. Our goal was to develop a longitudinally-aware segmentation network (LAS-Net) that can quantify serial PET/CT images for pediatric Hodgkin lymphoma patients. $\textbf{Materials and Methods}$: This retrospective study included baseline (PET1) and interim (PET2) PET/CT images from 297 patients enrolled in two Children's Oncology Group clinical trials (AHOD1331 and AHOD0831). LAS-Net incorporates longitudinal cross-attention, allowing relevant features from PET1 to inform the analysis of PET2. Model performance was evaluated using Dice coefficients for PET1 and detection F1 scores for PET2. Additionally, we extracted and compared quantitative PET metrics, including metabolic tumor volume (MTV) and total lesion glycolysis (TLG) in PET1, as well as qPET and $\Delta$SUVmax in PET2, against physician measurements. We quantified their agreement using Spearman's $\rho$ correlations and employed bootstrap resampling for statistical analysis. $\textbf{Results}$: LAS-Net detected residual lymphoma in PET2 with an F1 score of 0.606 (precision/recall: 0.615/0.600), outperforming all comparator methods (P<0.01). For baseline segmentation, LAS-Net achieved a mean Dice score of 0.772. In PET quantification, LAS-Net's measurements of qPET, $\Delta$SUVmax, MTV and TLG were strongly correlated with physician measurements, with Spearman's $\rho$ of 0.78, 0.80, 0.93 and 0.96, respectively. The performance remained high, with a slight decrease, in an external testing cohort. $\textbf{Conclusion}$: LAS-Net achieved high performance in quantifying PET metrics across serial scans, highlighting the value of longitudinal awareness in evaluating multi-time-point imaging datasets.