 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge$ abla^2$DFT: A Universal Quantum Chemistry Dataset of Drug-Like Molecules and a Benchmark for Neural Network Potentials
Jun 20, 2024Methods of computational quantum chemistry provide accurate approximations of molecular properties crucial for computer-aided drug discovery and other areas of chemical science. However, high computational complexity limits the scalability of their applications. Neural network potentials (NNPs) are a promising alternative to quantum chemistry methods, but they require large and diverse datasets for training. This work presents a new dataset and benchmark called $\nabla^2$DFT that is based on the nablaDFT. It contains twice as much molecular structures, three times more conformations, new data types and tasks, and state-of-the-art models. The dataset includes energies, forces, 17 molecular properties, Hamiltonian and overlap matrices, and a wavefunction object. All calculations were performed at the DFT level ($\omega$B97X-D/def2-SVP) for each conformation. Moreover, $\nabla^2$DFT is the first dataset that contains relaxation trajectories for a substantial number of drug-like molecules. We also introduce a novel benchmark for evaluating NNPs in molecular property prediction, Hamiltonian prediction, and conformational optimization tasks. Finally, we propose an extendable framework for training NNPs and implement 10 models within it.
FREED++: Improving RL Agents for Fragment-Based Molecule Generation by Thorough Reproduction
Jan 18, 2024



A rational design of new therapeutic drugs aims to find a molecular structure with desired biological functionality, e.g., an ability to activate or suppress a specific protein via binding to it. Molecular docking is a common technique for evaluating protein-molecule interactions. Recently, Reinforcement Learning (RL) has emerged as a promising approach to generating molecules with the docking score (DS) as a reward. In this work, we reproduce, scrutinize and improve the recent RL model for molecule generation called FREED (arXiv:2110.01219). Extensive evaluation of the proposed method reveals several limitations and challenges despite the outstanding results reported for three target proteins. Our contributions include fixing numerous implementation bugs and simplifying the model while increasing its quality, significantly extending experiments, and conducting an accurate comparison with current state-of-the-art methods for protein-conditioned molecule generation. We show that the resulting fixed model is capable of producing molecules with superior docking scores compared to alternative approaches.
Gradual Optimization Learning for Conformational Energy Minimization
Nov 05, 2023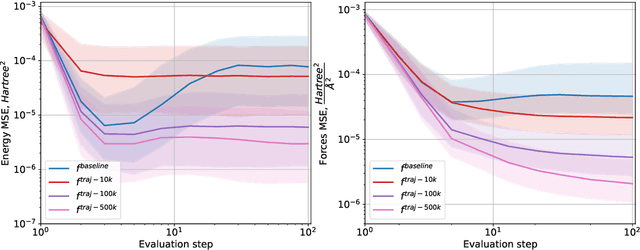

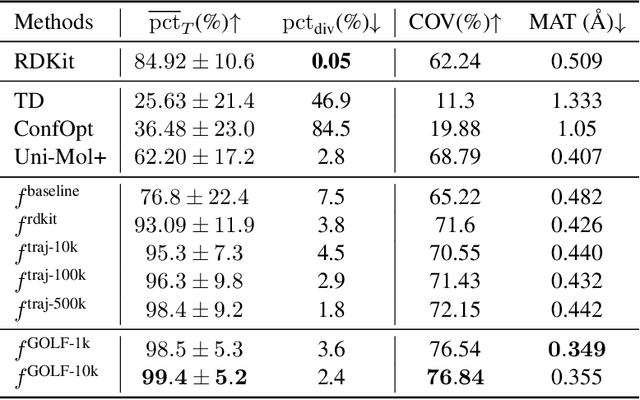
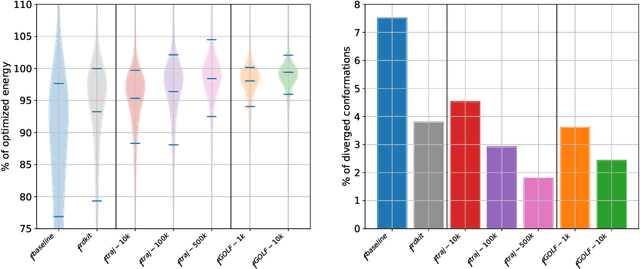
Molecular conformation optimization is crucial to computer-aided drug discovery and materials design. Traditional energy minimization techniques rely on iterative optimization methods that use molecular forces calculated by a physical simulator (oracle) as anti-gradients. However, this is a computationally expensive approach that requires many interactions with a physical simulator. One way to accelerate this procedure is to replace the physical simulator with a neural network. Despite recent progress in neural networks for molecular conformation energy prediction, such models are prone to distribution shift, leading to inaccurate energy minimization. We find that the quality of energy minimization with neural networks can be improved by providing optimization trajectories as additional training data. Still, it takes around $5 \times 10^5$ additional conformations to match the physical simulator's optimization quality. In this work, we present the Gradual Optimization Learning Framework (GOLF) for energy minimization with neural networks that significantly reduces the required additional data. The framework consists of an efficient data-collecting scheme and an external optimizer. The external optimizer utilizes gradients from the energy prediction model to generate optimization trajectories, and the data-collecting scheme selects additional training data to be processed by the physical simulator. Our results demonstrate that the neural network trained with GOLF performs on par with the oracle on a benchmark of diverse drug-like molecules using $50$x less additional data.
Drug and Disease Interpretation Learning with Biomedical Entity Representation Transformer
Jan 22, 2021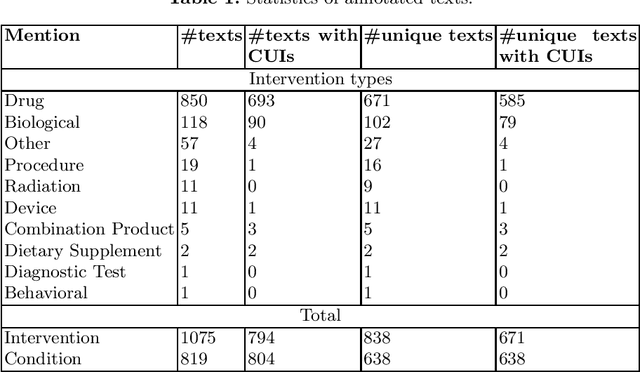
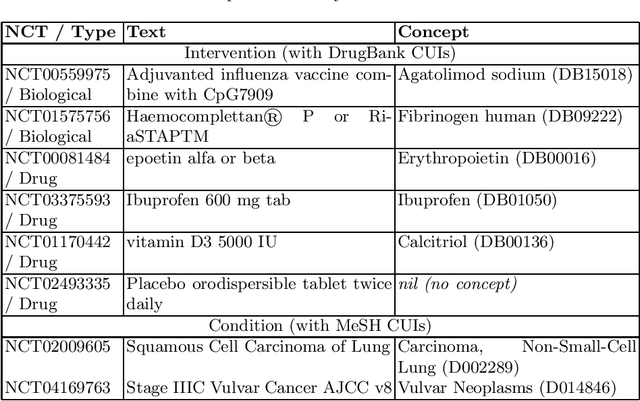
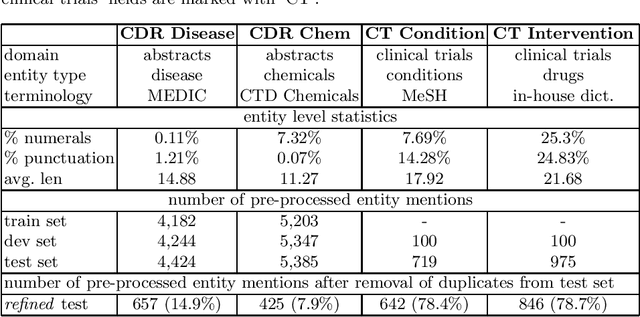
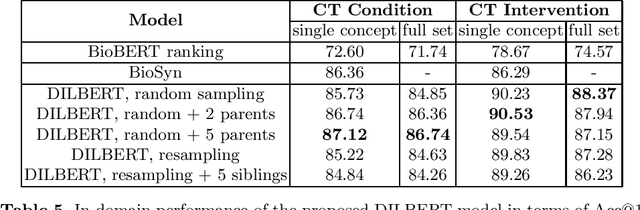
Concept normalization in free-form texts is a crucial step in every text-mining pipeline. Neural architectures based on Bidirectional Encoder Representations from Transformers (BERT) have achieved state-of-the-art results in the biomedical domain. In the context of drug discovery and development, clinical trials are necessary to establish the efficacy and safety of drugs. We investigate the effectiveness of transferring concept normalization from the general biomedical domain to the clinical trials domain in a zero-shot setting with an absence of labeled data. We propose a simple and effective two-stage neural approach based on fine-tuned BERT architectures. In the first stage, we train a metric learning model that optimizes relative similarity of mentions and concepts via triplet loss. The model is trained on available labeled corpora of scientific abstracts to obtain vector embeddings of concept names and entity mentions from texts. In the second stage, we find the closest concept name representation in an embedding space to a given clinical mention. We evaluated several models, including state-of-the-art architectures, on a dataset of abstracts and a real-world dataset of trial records with interventions and conditions mapped to drug and disease terminologies. Extensive experiments validate the effectiveness of our approach in knowledge transfer from the scientific literature to clinical trials.
Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models
Nov 29, 2018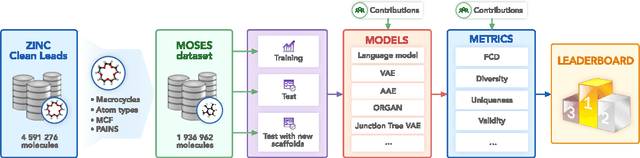


Deep generative models such as generative adversarial networks, variational autoencoders, and autoregressive models are rapidly growing in popularity for the discovery of new molecules and materials. In this work, we introduce MOlecular SEtS (MOSES), a benchmarking platform to support research on machine learning for drug discovery. MOSES implements several popular molecular generation models and includes a set of metrics that evaluate the diversity and quality of generated molecules. MOSES is meant to standardize the research on the molecular generation and facilitate the sharing and comparison of new models. Additionally, we provide a large-scale comparison of existing state of the art models and elaborate on current challenges for generative models that might prove fertile ground for new research. Our platform and source code are freely available at https://github.com/molecularsets/