 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Generalized Genetic Random Field Method for the Genetic Association Analysis of Sequencing Data
Aug 18, 2025With the advance of high-throughput sequencing technologies, it has become feasible to investigate the influence of the entire spectrum of sequencing variations on complex human diseases. Although association studies utilizing the new sequencing technologies hold great promise to unravel novel genetic variants, especially rare genetic variants that contribute to human diseases, the statistical analysis of high-dimensional sequencing data remains a challenge. Advanced analytical methods are in great need to facilitate high-dimensional sequencing data analyses. In this article, we propose a generalized genetic random field (GGRF) method for association analyses of sequencing data. Like other similarity-based methods (e.g., SIMreg and SKAT), the new method has the advantages of avoiding the need to specify thresholds for rare variants and allowing for testing multiple variants acting in different directions and magnitude of effects. The method is built on the generalized estimating equation framework and thus accommodates a variety of disease phenotypes (e.g., quantitative and binary phenotypes). Moreover, it has a nice asymptotic property, and can be applied to small-scale sequencing data without need for small-sample adjustment. Through simulations, we demonstrate that the proposed GGRF attains an improved or comparable power over a commonly used method, SKAT, under various disease scenarios, especially when rare variants play a significant role in disease etiology. We further illustrate GGRF with an application to a real dataset from the Dallas Heart Study. By using GGRF, we were able to detect the association of two candidate genes, ANGPTL3 and ANGPTL4, with serum triglyceride.
Repurposing Foundation Model for Generalizable Medical Time Series Classification
Oct 03, 2024



Medical time series (MedTS) classification is critical for a wide range of healthcare applications such as Alzheimer's Disease diagnosis. However, its real-world deployment is severely challenged by poor generalizability due to inter- and intra-dataset heterogeneity in MedTS, including variations in channel configurations, time series lengths, and diagnostic tasks. Here, we propose FORMED, a foundation classification model that leverages a pre-trained backbone and tackles these challenges through re-purposing. FORMED integrates the general representation learning enabled by the backbone foundation model and the medical domain knowledge gained on a curated cohort of MedTS datasets. FORMED can adapt seamlessly to unseen MedTS datasets, regardless of the number of channels, sample lengths, or medical tasks. Experimental results show that, without any task-specific adaptation, the repurposed FORMED achieves performance that is competitive with, and often superior to, 11 baseline models trained specifically for each dataset. Furthermore, FORMED can effectively adapt to entirely new, unseen datasets, with lightweight parameter updates, consistently outperforming baselines. Our results highlight FORMED as a versatile and scalable model for a wide range of MedTS classification tasks, positioning it as a strong foundation model for future research in MedTS analysis.
Ensembling improves stability and power of feature selection for deep learning models
Oct 02, 2022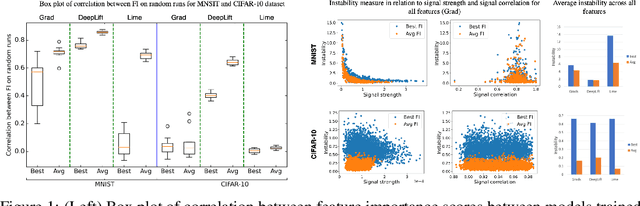
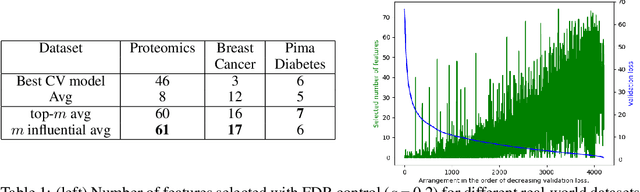
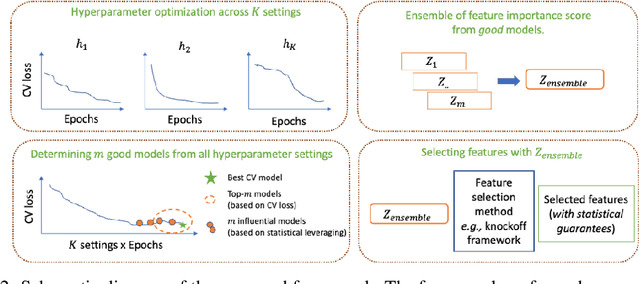
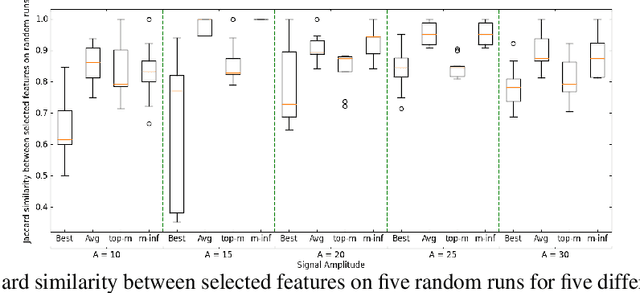
With the growing adoption of deep learning models in different real-world domains, including computational biology, it is often necessary to understand which data features are essential for the model's decision. Despite extensive recent efforts to define different feature importance metrics for deep learning models, we identified that inherent stochasticity in the design and training of deep learning models makes commonly used feature importance scores unstable. This results in varied explanations or selections of different features across different runs of the model. We demonstrate how the signal strength of features and correlation among features directly contribute to this instability. To address this instability, we explore the ensembling of feature importance scores of models across different epochs and find that this simple approach can substantially address this issue. For example, we consider knockoff inference as they allow feature selection with statistical guarantees. We discover considerable variability in selected features in different epochs of deep learning training, and the best selection of features doesn't necessarily occur at the lowest validation loss, the conventional approach to determine the best model. As such, we present a framework to combine the feature importance of trained models across different hyperparameter settings and epochs, and instead of selecting features from one best model, we perform an ensemble of feature importance scores from numerous good models. Across the range of experiments in simulated and various real-world datasets, we demonstrate that the proposed framework consistently improves the power of feature selection.
Improving genetic risk prediction across diverse population by disentangling ancestry representations
May 10, 2022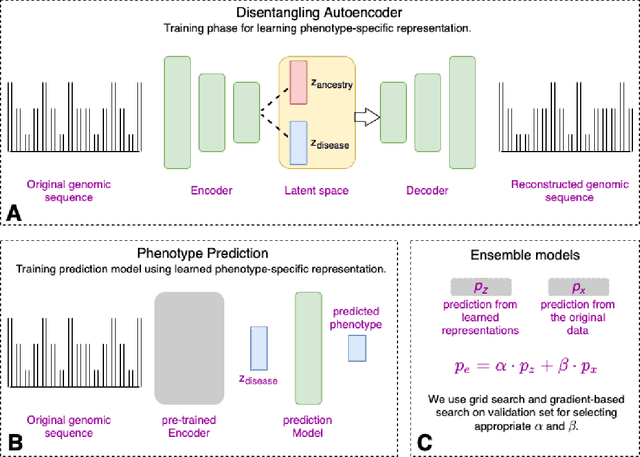
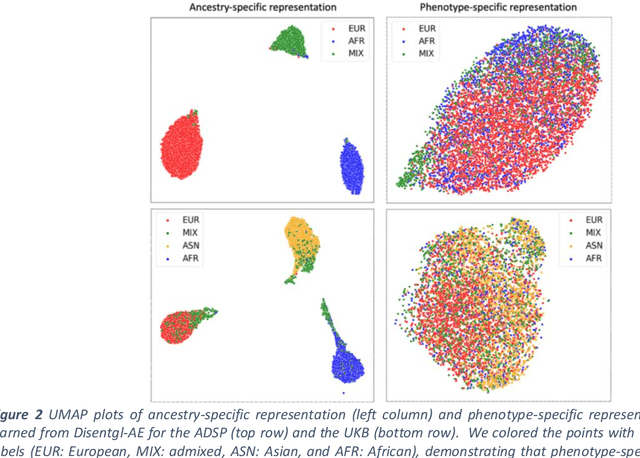
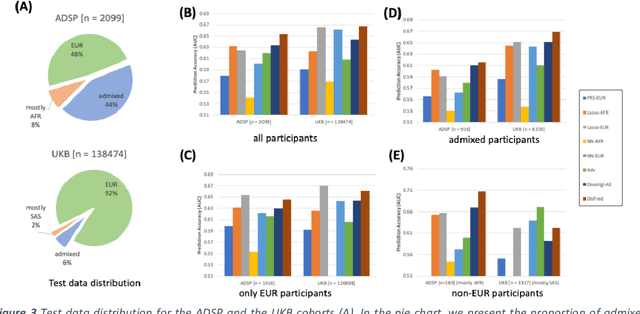
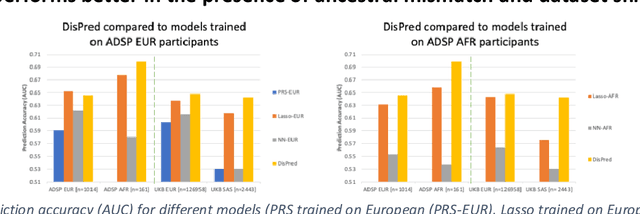
Risk prediction models using genetic data have seen increasing traction in genomics. However, most of the polygenic risk models were developed using data from participants with similar (mostly European) ancestry. This can lead to biases in the risk predictors resulting in poor generalization when applied to minority populations and admixed individuals such as African Americans. To address this bias, largely due to the prediction models being confounded by the underlying population structure, we propose a novel deep-learning framework that leverages data from diverse population and disentangles ancestry from the phenotype-relevant information in its representation. The ancestry disentangled representation can be used to build risk predictors that perform better across minority populations. We applied the proposed method to the analysis of Alzheimer's disease genetics. Comparing with standard linear and nonlinear risk prediction methods, the proposed method substantially improves risk prediction in minority populations, particularly for admixed individuals.
Deep neural networks with controlled variable selection for the identification of putative causal genetic variants
Sep 29, 2021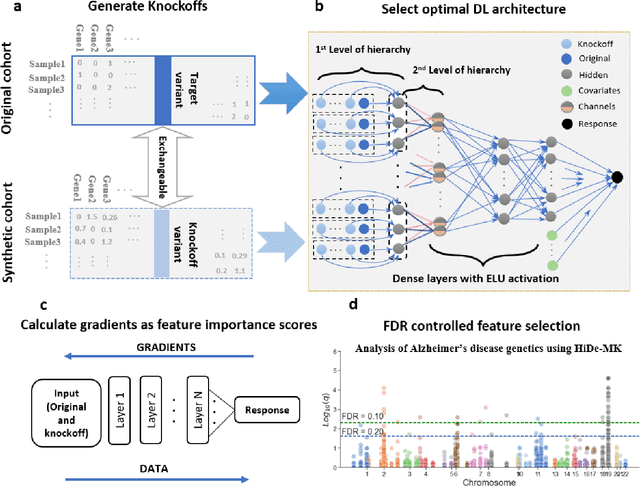
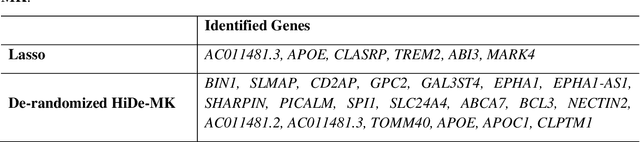
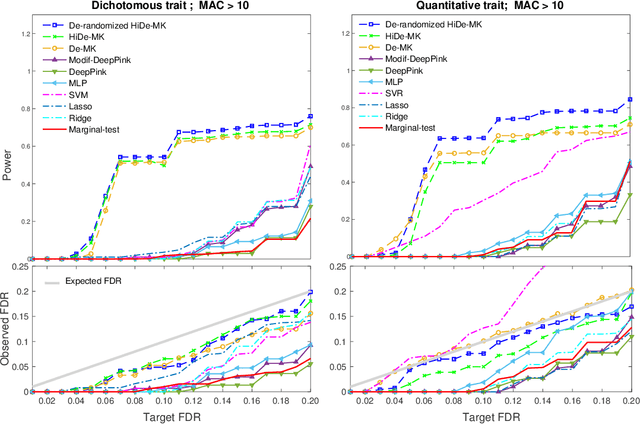
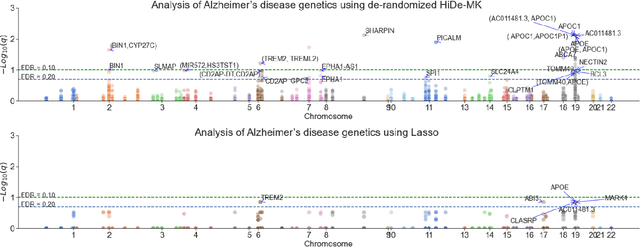
Deep neural networks (DNN) have been used successfully in many scientific problems for their high prediction accuracy, but their application to genetic studies remains challenging due to their poor interpretability. In this paper, we consider the problem of scalable, robust variable selection in DNN for the identification of putative causal genetic variants in genome sequencing studies. We identified a pronounced randomness in feature selection in DNN due to its stochastic nature, which may hinder interpretability and give rise to misleading results. We propose an interpretable neural network model, stabilized using ensembling, with controlled variable selection for genetic studies. The merit of the proposed method includes: (1) flexible modelling of the non-linear effect of genetic variants to improve statistical power; (2) multiple knockoffs in the input layer to rigorously control false discovery rate; (3) hierarchical layers to substantially reduce the number of weight parameters and activations to improve computational efficiency; (4) de-randomized feature selection to stabilize identified signals. We evaluated the proposed method in extensive simulation studies and applied it to the analysis of Alzheimer disease genetics. We showed that the proposed method, when compared to conventional linear and nonlinear methods, can lead to substantially more discoveries.