 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProvably Convergent Plug-and-play Proximal Block Coordinate Descent Method for Hyperspectral Anomaly Detection
Dec 19, 2024
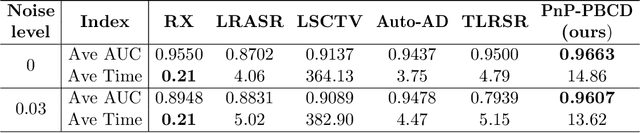
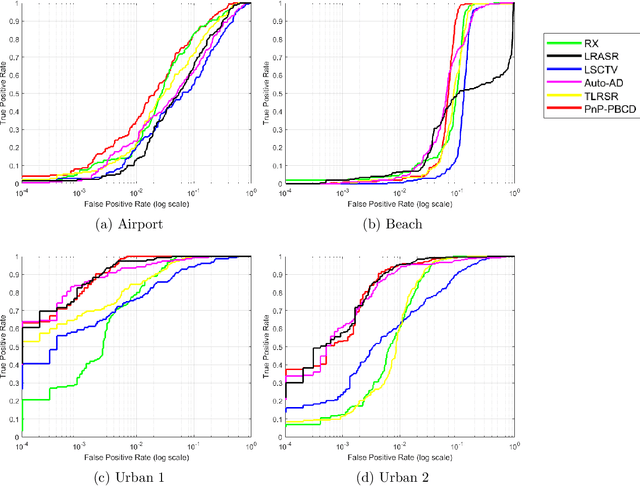
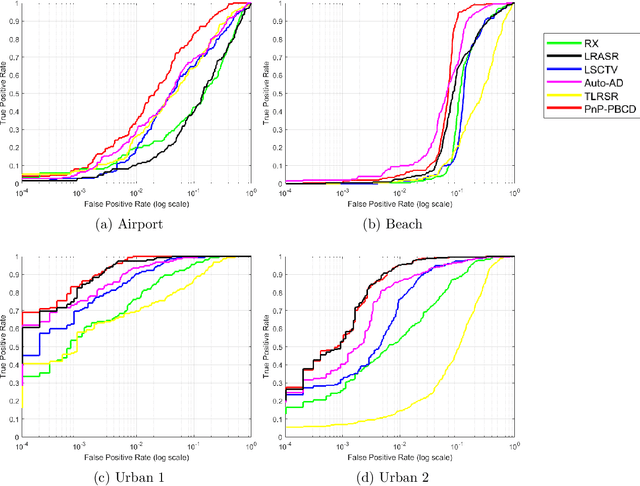
Hyperspectral anomaly detection refers to identifying pixels in the hyperspectral images that have spectral characteristics significantly different from the background. In this paper, we introduce a novel model that represents the background information using a low-rank representation. We integrate an implicit proximal denoiser prior, associated with a deep learning based denoiser, within a plug-and-play (PnP) framework to effectively remove noise from the eigenimages linked to the low-rank representation. Anomalies are characterized using a generalized group sparsity measure, denoted as $\|\cdot\|_{2,\psi}$. To solve the resulting orthogonal constrained nonconvex nonsmooth optimization problem, we develop a PnP-proximal block coordinate descent (PnP-PBCD) method, where the eigenimages are updated using a proximal denoiser within the PnP framework. We prove that any accumulation point of the sequence generated by the PnP-PBCD method is a stationary point. We evaluate the effectiveness of the PnP-PBCD method on hyperspectral anomaly detection in scenarios with and without Gaussian noise contamination. The results demonstrate that the proposed method can effectively detect anomalous objects, outperforming the competing methods that may mistakenly identify noise as anomalies or misidentify the anomalous objects due to noise interference.
Orthogonal Constrained Minimization with Tensor $\ell_{2,p}$ Regularization for HSI Denoising and Destriping
Jul 04, 2024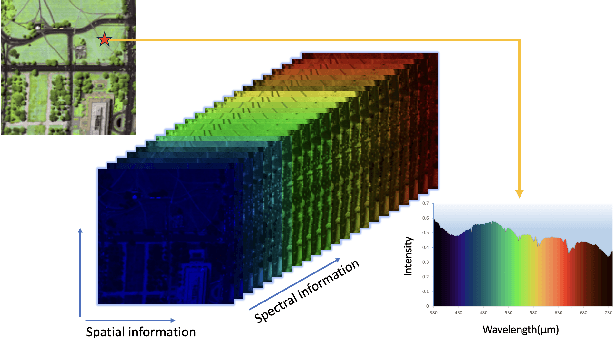
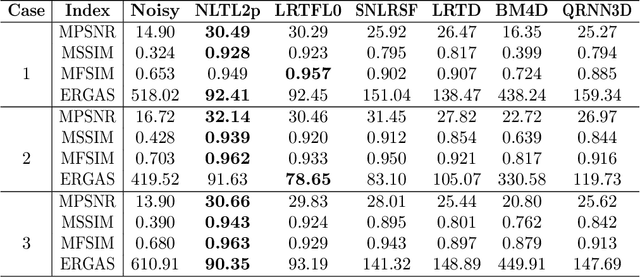
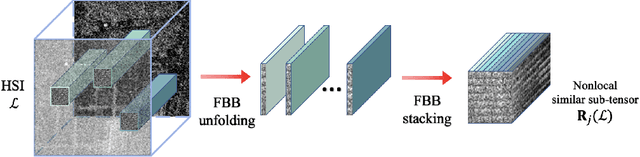
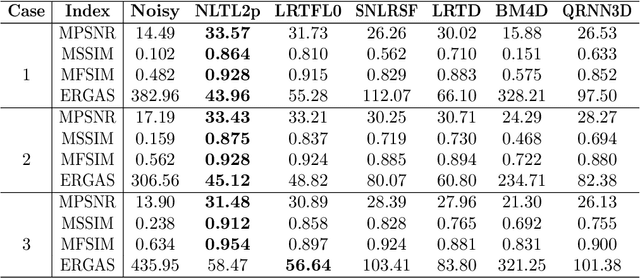
Hyperspectral images (HSIs) are often contaminated by a mixture of noises such as Gaussian noise, dead lines, stripes, and so on. In this paper, we propose a novel approach for HSI denoising and destriping, called NLTL2p, which consists of an orthogonal constrained minimization model and an iterative algorithm with convergence guarantees. The model of the proposed NLTL2p approach is built based on a new sparsity-enhanced Nonlocal Low-rank Tensor regularization and a tensor $\ell_{2,p}$ norm with $p\in(0,1)$. The low-rank constraints for HSI denoising utilize the spatial nonlocal self-similarity and spectral correlation of HSIs and are formulated based on independent higher-order singular value decomposition with sparsity enhancement on its core tensor to prompt more low-rankness. The tensor $\ell_{2,p}$ norm for HSI destriping is extended from the matrix $\ell_{2,p}$ norm. A proximal block coordinate descent algorithm is proposed in the NLTL2p approach to solve the resulting nonconvex nonsmooth minimization with orthogonal constraints. We show any accumulation point of the sequence generated by the proposed algorithm converges to a first-order stationary point, which is defined using three equalities of substationarity, symmetry, and feasibility for orthogonal constraints. In the numerical experiments, we compare the proposed method with state-of-the-art methods including a deep learning based method, and test the methods on both simulated and real HSI datasets. Our proposed NLTL2p method demonstrates outperformance in terms of metrics such as mean peak signal-to-noise ratio as well as visual quality.
Ensembling improves stability and power of feature selection for deep learning models
Oct 02, 2022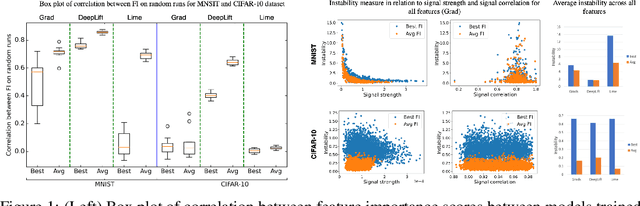
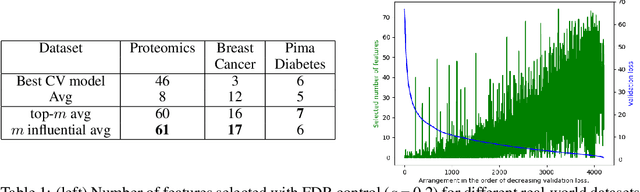
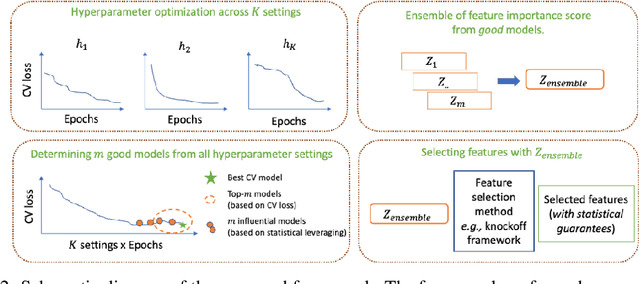
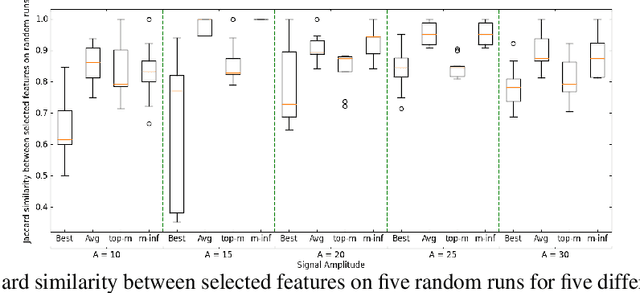
With the growing adoption of deep learning models in different real-world domains, including computational biology, it is often necessary to understand which data features are essential for the model's decision. Despite extensive recent efforts to define different feature importance metrics for deep learning models, we identified that inherent stochasticity in the design and training of deep learning models makes commonly used feature importance scores unstable. This results in varied explanations or selections of different features across different runs of the model. We demonstrate how the signal strength of features and correlation among features directly contribute to this instability. To address this instability, we explore the ensembling of feature importance scores of models across different epochs and find that this simple approach can substantially address this issue. For example, we consider knockoff inference as they allow feature selection with statistical guarantees. We discover considerable variability in selected features in different epochs of deep learning training, and the best selection of features doesn't necessarily occur at the lowest validation loss, the conventional approach to determine the best model. As such, we present a framework to combine the feature importance of trained models across different hyperparameter settings and epochs, and instead of selecting features from one best model, we perform an ensemble of feature importance scores from numerous good models. Across the range of experiments in simulated and various real-world datasets, we demonstrate that the proposed framework consistently improves the power of feature selection.
Improving genetic risk prediction across diverse population by disentangling ancestry representations
May 10, 2022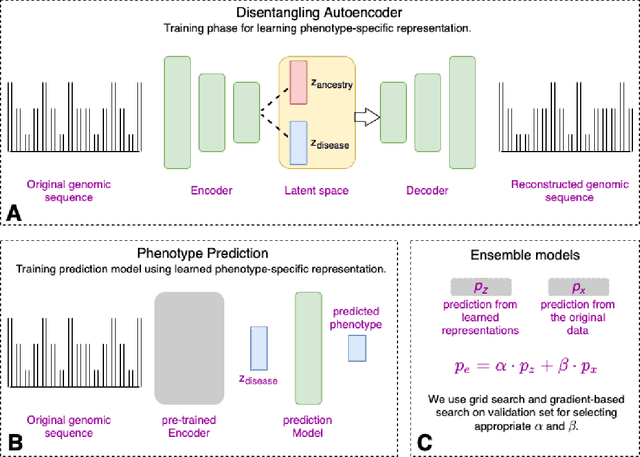
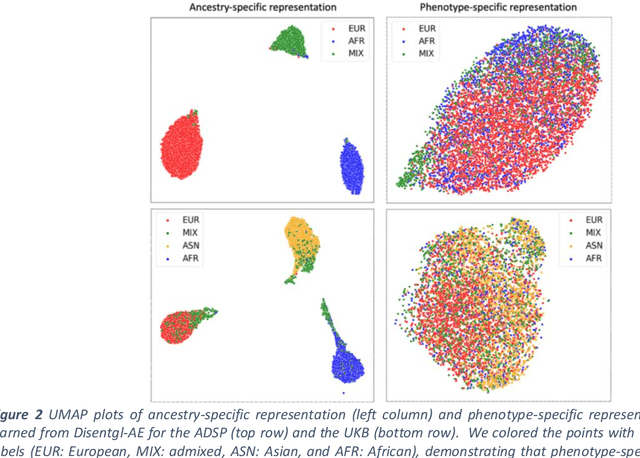
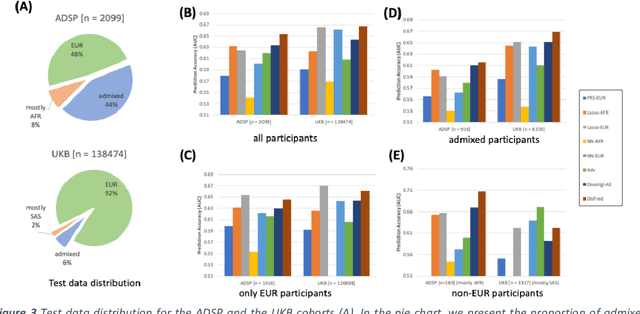
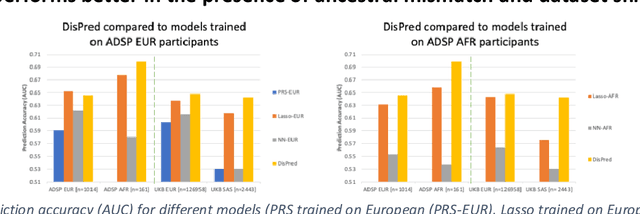
Risk prediction models using genetic data have seen increasing traction in genomics. However, most of the polygenic risk models were developed using data from participants with similar (mostly European) ancestry. This can lead to biases in the risk predictors resulting in poor generalization when applied to minority populations and admixed individuals such as African Americans. To address this bias, largely due to the prediction models being confounded by the underlying population structure, we propose a novel deep-learning framework that leverages data from diverse population and disentangles ancestry from the phenotype-relevant information in its representation. The ancestry disentangled representation can be used to build risk predictors that perform better across minority populations. We applied the proposed method to the analysis of Alzheimer's disease genetics. Comparing with standard linear and nonlinear risk prediction methods, the proposed method substantially improves risk prediction in minority populations, particularly for admixed individuals.