 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving genetic risk prediction across diverse population by disentangling ancestry representations
May 10, 2022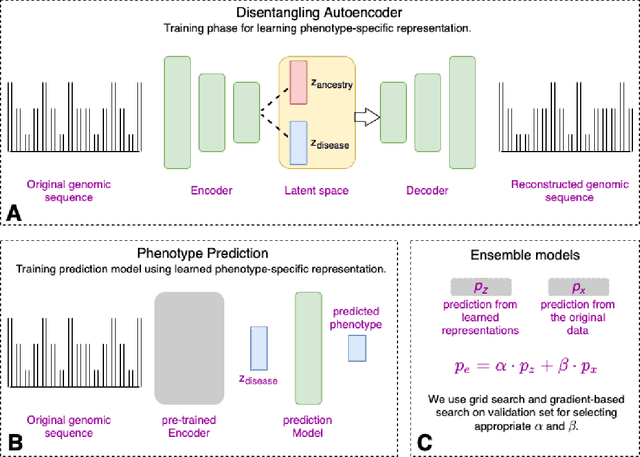
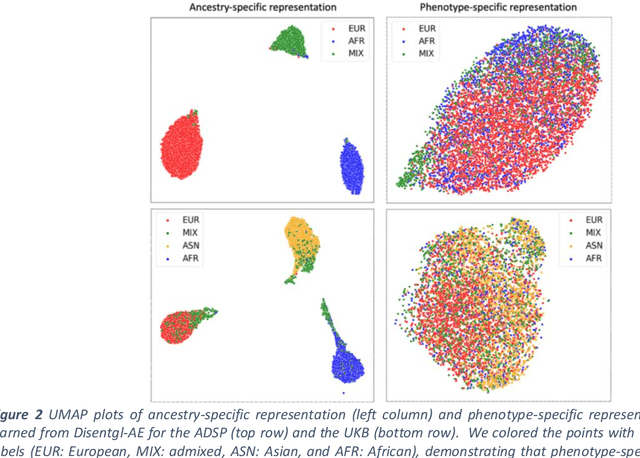
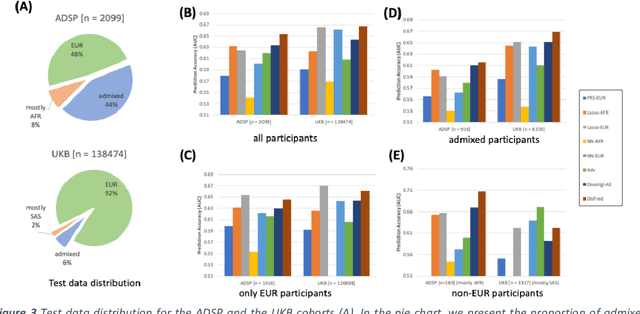
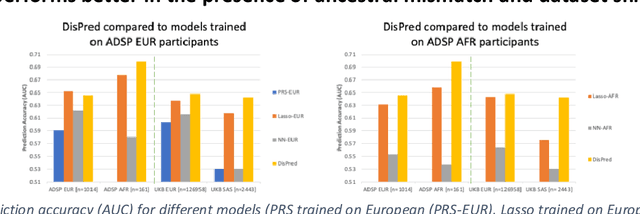
Risk prediction models using genetic data have seen increasing traction in genomics. However, most of the polygenic risk models were developed using data from participants with similar (mostly European) ancestry. This can lead to biases in the risk predictors resulting in poor generalization when applied to minority populations and admixed individuals such as African Americans. To address this bias, largely due to the prediction models being confounded by the underlying population structure, we propose a novel deep-learning framework that leverages data from diverse population and disentangles ancestry from the phenotype-relevant information in its representation. The ancestry disentangled representation can be used to build risk predictors that perform better across minority populations. We applied the proposed method to the analysis of Alzheimer's disease genetics. Comparing with standard linear and nonlinear risk prediction methods, the proposed method substantially improves risk prediction in minority populations, particularly for admixed individuals.
Deep neural networks with controlled variable selection for the identification of putative causal genetic variants
Sep 29, 2021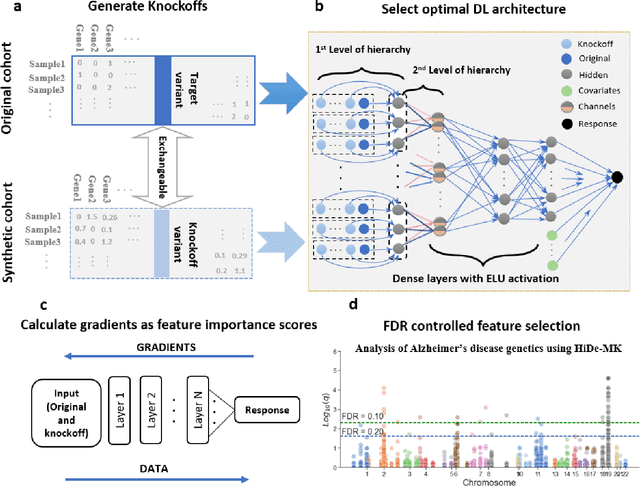
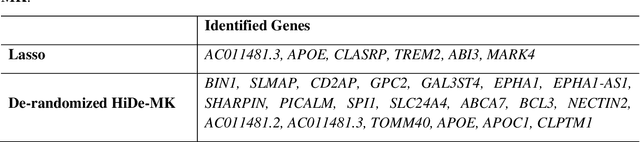
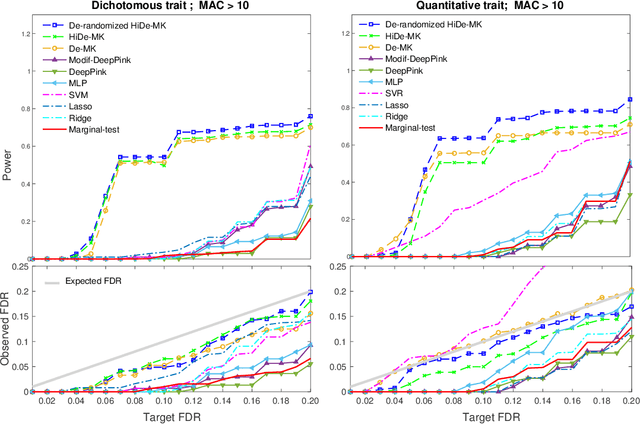
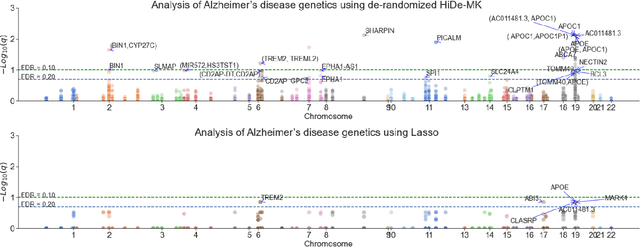
Deep neural networks (DNN) have been used successfully in many scientific problems for their high prediction accuracy, but their application to genetic studies remains challenging due to their poor interpretability. In this paper, we consider the problem of scalable, robust variable selection in DNN for the identification of putative causal genetic variants in genome sequencing studies. We identified a pronounced randomness in feature selection in DNN due to its stochastic nature, which may hinder interpretability and give rise to misleading results. We propose an interpretable neural network model, stabilized using ensembling, with controlled variable selection for genetic studies. The merit of the proposed method includes: (1) flexible modelling of the non-linear effect of genetic variants to improve statistical power; (2) multiple knockoffs in the input layer to rigorously control false discovery rate; (3) hierarchical layers to substantially reduce the number of weight parameters and activations to improve computational efficiency; (4) de-randomized feature selection to stabilize identified signals. We evaluated the proposed method in extensive simulation studies and applied it to the analysis of Alzheimer disease genetics. We showed that the proposed method, when compared to conventional linear and nonlinear methods, can lead to substantially more discoveries.