 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBanditLP: Large-Scale Stochastic Optimization for Personalized Recommendations
Jan 22, 2026We present BanditLP, a scalable multi-stakeholder contextual bandit framework that unifies neural Thompson Sampling for learning objective-specific outcomes with a large-scale linear program for constrained action selection at serving time. The methodology is application-agnostic, compatible with arbitrary neural architectures, and deployable at web scale, with an LP solver capable of handling billions of variables. Experiments on public benchmarks and synthetic data show consistent gains over strong baselines. We apply this approach in LinkedIn's email marketing system and demonstrate business win, illustrating the value of integrated exploration and constrained optimization in production.
A Generalized Genetic Random Field Method for the Genetic Association Analysis of Sequencing Data
Aug 18, 2025With the advance of high-throughput sequencing technologies, it has become feasible to investigate the influence of the entire spectrum of sequencing variations on complex human diseases. Although association studies utilizing the new sequencing technologies hold great promise to unravel novel genetic variants, especially rare genetic variants that contribute to human diseases, the statistical analysis of high-dimensional sequencing data remains a challenge. Advanced analytical methods are in great need to facilitate high-dimensional sequencing data analyses. In this article, we propose a generalized genetic random field (GGRF) method for association analyses of sequencing data. Like other similarity-based methods (e.g., SIMreg and SKAT), the new method has the advantages of avoiding the need to specify thresholds for rare variants and allowing for testing multiple variants acting in different directions and magnitude of effects. The method is built on the generalized estimating equation framework and thus accommodates a variety of disease phenotypes (e.g., quantitative and binary phenotypes). Moreover, it has a nice asymptotic property, and can be applied to small-scale sequencing data without need for small-sample adjustment. Through simulations, we demonstrate that the proposed GGRF attains an improved or comparable power over a commonly used method, SKAT, under various disease scenarios, especially when rare variants play a significant role in disease etiology. We further illustrate GGRF with an application to a real dataset from the Dallas Heart Study. By using GGRF, we were able to detect the association of two candidate genes, ANGPTL3 and ANGPTL4, with serum triglyceride.
Causal Predictive Optimization and Generation for Business AI
May 14, 2025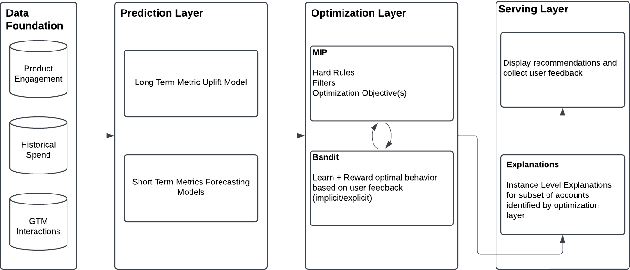


The sales process involves sales functions converting leads or opportunities to customers and selling more products to existing customers. The optimization of the sales process thus is key to success of any B2B business. In this work, we introduce a principled approach to sales optimization and business AI, namely the Causal Predictive Optimization and Generation, which includes three layers: 1) prediction layer with causal ML 2) optimization layer with constraint optimization and contextual bandit 3) serving layer with Generative AI and feedback-loop for system enhancement. We detail the implementation and deployment of the system in LinkedIn, showcasing significant wins over legacy systems and sharing learning and insight broadly applicable to this field.
LiDDA: Data Driven Attribution at LinkedIn
May 14, 2025Data Driven Attribution, which assigns conversion credits to marketing interactions based on causal patterns learned from data, is the foundation of modern marketing intelligence and vital to any marketing businesses and advertising platform. In this paper, we introduce a unified transformer-based attribution approach that can handle member-level data, aggregate-level data, and integration of external macro factors. We detail the large scale implementation of the approach at LinkedIn, showcasing significant impact. We also share learning and insights that are broadly applicable to the marketing and ad tech fields.
Beyond Basic A/B testing: Improving Statistical Efficiency for Business Growth
May 13, 2025The standard A/B testing approaches are mostly based on t-test in large scale industry applications. These standard approaches however suffers from low statistical power in business settings, due to nature of small sample-size or non-Gaussian distribution or return-on-investment (ROI) consideration. In this paper, we propose several approaches to addresses these challenges: (i) regression adjustment, generalized estimating equation, Man-Whitney U and Zero-Trimmed U that addresses each of these issues separately, and (ii) a novel doubly robust generalized U that handles ROI consideration, distribution robustness and small samples in one framework. We provide theoretical results on asymptotic normality and efficiency bounds, together with insights on the efficiency gain from theoretical analysis. We further conduct comprehensive simulation studies and apply the methods to multiple real A/B tests.
Neural Optimization with Adaptive Heuristics for Intelligent Marketing System
May 17, 2024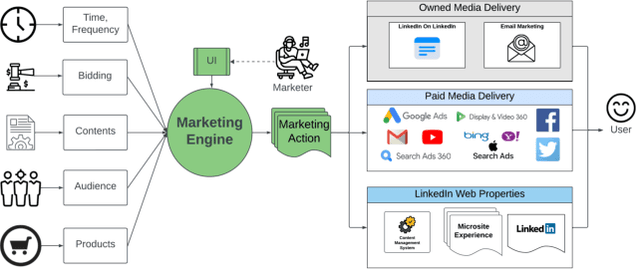

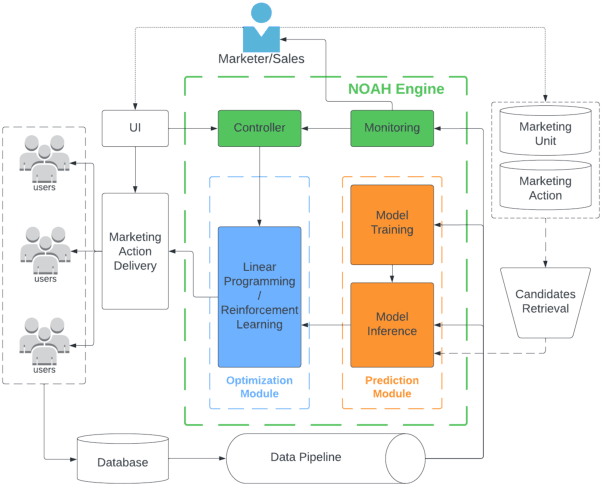

Computational marketing has become increasingly important in today's digital world, facing challenges such as massive heterogeneous data, multi-channel customer journeys, and limited marketing budgets. In this paper, we propose a general framework for marketing AI systems, the Neural Optimization with Adaptive Heuristics (NOAH) framework. NOAH is the first general framework for marketing optimization that considers both to-business (2B) and to-consumer (2C) products, as well as both owned and paid channels. We describe key modules of the NOAH framework, including prediction, optimization, and adaptive heuristics, providing examples for bidding and content optimization. We then detail the successful application of NOAH to LinkedIn's email marketing system, showcasing significant wins over the legacy ranking system. Additionally, we share details and insights that are broadly useful, particularly on: (i) addressing delayed feedback with lifetime value, (ii) performing large-scale linear programming with randomization, (iii) improving retrieval with audience expansion, (iv) reducing signal dilution in targeting tests, and (v) handling zero-inflated heavy-tail metrics in statistical testing.
Generalized Similarity U: A Non-parametric Test of Association Based on Similarity
Jan 04, 2018
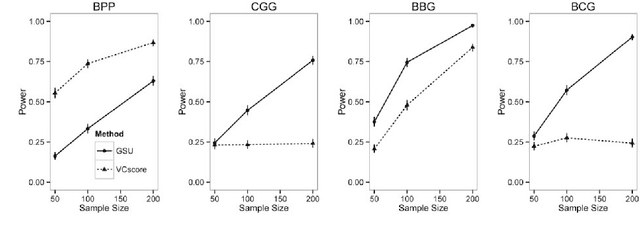
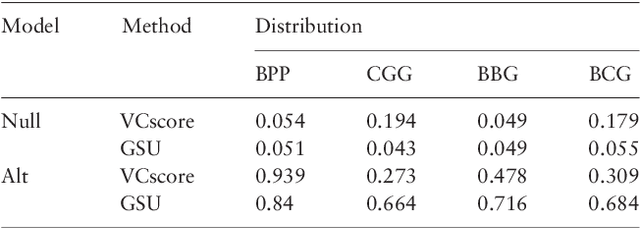

Second generation sequencing technologies are being increasingly used for genetic association studies, where the main research interest is to identify sets of genetic variants that contribute to various phenotype. The phenotype can be univariate disease status, multivariate responses and even high-dimensional outcomes. Considering the genotype and phenotype as two complex objects, this also poses a general statistical problem of testing association between complex objects. We here proposed a similarity-based test, generalized similarity U (GSU), that can test the association between complex objects. We first studied the theoretical properties of the test in a general setting and then focused on the application of the test to sequencing association studies. Based on theoretical analysis, we proposed to use Laplacian kernel based similarity for GSU to boost power and enhance robustness. Through simulation, we found that GSU did have advantages over existing methods in terms of power and robustness. We further performed a whole genome sequencing (WGS) scan for Alzherimer Disease Neuroimaging Initiative (ADNI) data, identifying three genes, APOE, APOC1 and TOMM40, associated with imaging phenotype. We developed a C++ package for analysis of whole genome sequencing data using GSU. The source codes can be downloaded at https://github.com/changshuaiwei/gsu.
Trees Assembling Mann Whitney Approach for Detecting Genome-wide Joint Association among Low Marginal Effect loci
May 05, 2015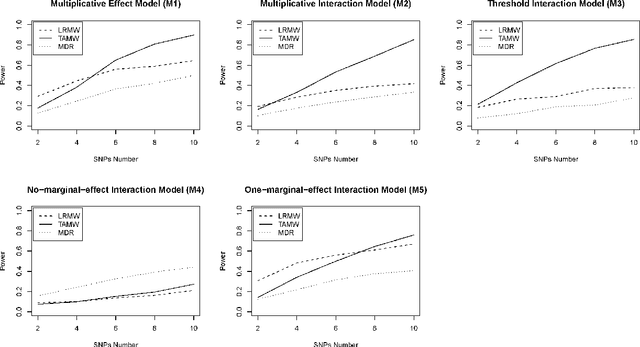
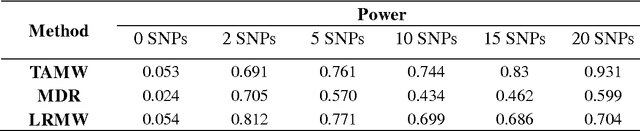

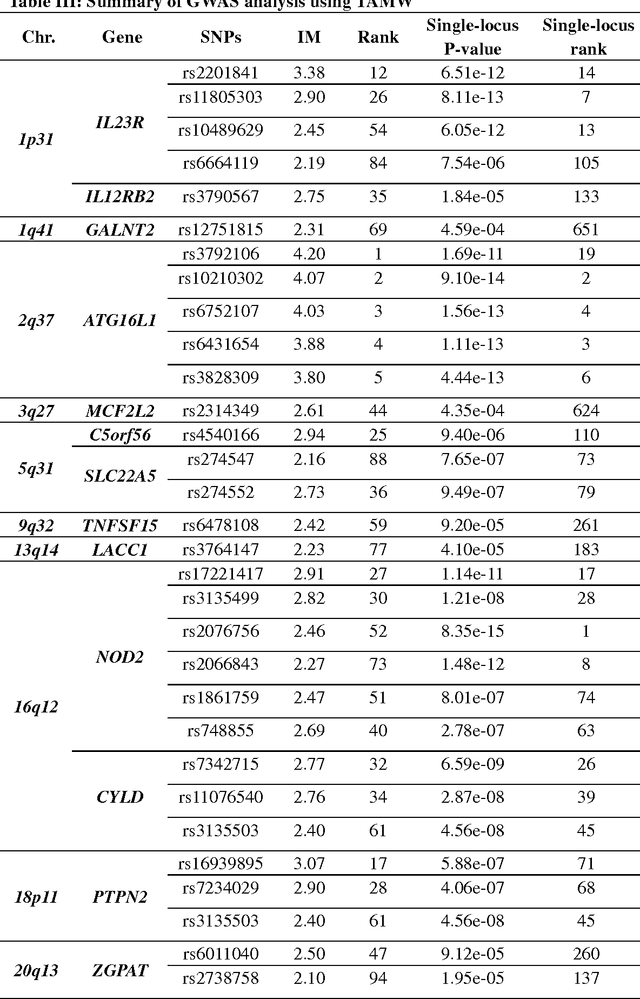
Common complex diseases are likely influenced by the interplay of hundreds, or even thousands, of genetic variants. Converging evidence shows that genetic variants with low marginal effects (LME) play an important role in disease development. Despite their potential significance, discovering LME genetic variants and assessing their joint association on high dimensional data (e.g., genome wide association studies) remain a great challenge. To facilitate joint association analysis among a large ensemble of LME genetic variants, we proposed a computationally efficient and powerful approach, which we call Trees Assembling Mann whitney (TAMW). Through simulation studies and an empirical data application, we found that TAMW outperformed multifactor dimensionality reduction (MDR) and the likelihood ratio based Mann whitney approach (LRMW) when the underlying complex disease involves multiple LME loci and their interactions. For instance, in a simulation with 20 interacting LME loci, TAMW attained a higher power (power=0.931) than both MDR (power=0.599) and LRMW (power=0.704). In an empirical study of 29 known Crohn's disease (CD) loci, TAMW also identified a stronger joint association with CD than those detected by MDR and LRMW. Finally, we applied TAMW to Wellcome Trust CD GWAS to conduct a genome wide analysis. The analysis of 459K single nucleotide polymorphisms was completed in 40 hours using parallel computing, and revealed a joint association predisposing to CD (p-value=2.763e-19). Further analysis of the newly discovered association suggested that 13 genes, such as ATG16L1 and LACC1, may play an important role in CD pathophysiological and etiological processes.