 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTrees Assembling Mann Whitney Approach for Detecting Genome-wide Joint Association among Low Marginal Effect loci
Paper and Code
May 05, 2015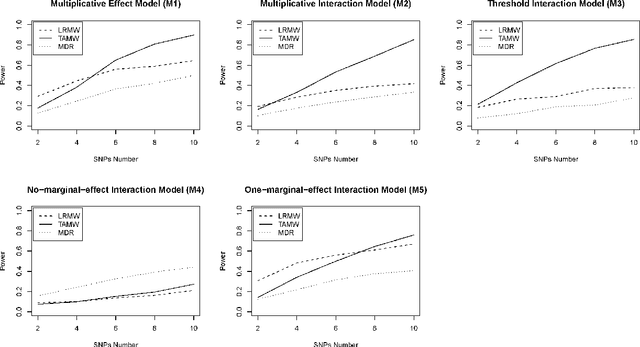
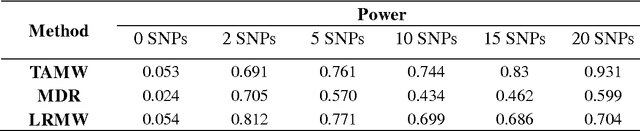

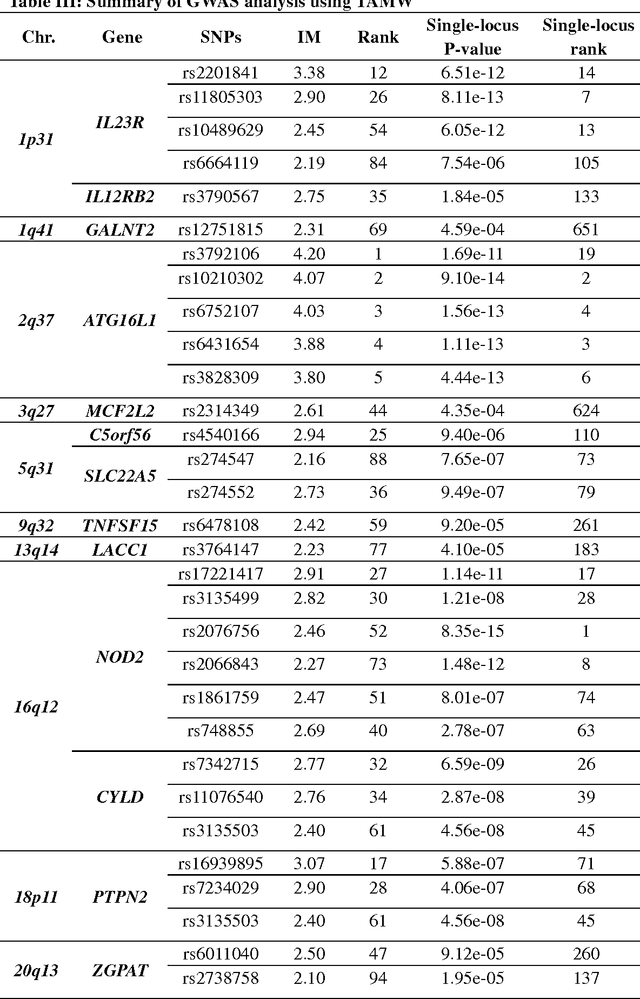
Common complex diseases are likely influenced by the interplay of hundreds, or even thousands, of genetic variants. Converging evidence shows that genetic variants with low marginal effects (LME) play an important role in disease development. Despite their potential significance, discovering LME genetic variants and assessing their joint association on high dimensional data (e.g., genome wide association studies) remain a great challenge. To facilitate joint association analysis among a large ensemble of LME genetic variants, we proposed a computationally efficient and powerful approach, which we call Trees Assembling Mann whitney (TAMW). Through simulation studies and an empirical data application, we found that TAMW outperformed multifactor dimensionality reduction (MDR) and the likelihood ratio based Mann whitney approach (LRMW) when the underlying complex disease involves multiple LME loci and their interactions. For instance, in a simulation with 20 interacting LME loci, TAMW attained a higher power (power=0.931) than both MDR (power=0.599) and LRMW (power=0.704). In an empirical study of 29 known Crohn's disease (CD) loci, TAMW also identified a stronger joint association with CD than those detected by MDR and LRMW. Finally, we applied TAMW to Wellcome Trust CD GWAS to conduct a genome wide analysis. The analysis of 459K single nucleotide polymorphisms was completed in 40 hours using parallel computing, and revealed a joint association predisposing to CD (p-value=2.763e-19). Further analysis of the newly discovered association suggested that 13 genes, such as ATG16L1 and LACC1, may play an important role in CD pathophysiological and etiological processes.