 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePushing the Limits of All-Atom Geometric Graph Neural Networks: Pre-Training, Scaling and Zero-Shot Transfer
Oct 29, 2024Constructing transferable descriptors for conformation representation of molecular and biological systems finds numerous applications in drug discovery, learning-based molecular dynamics, and protein mechanism analysis. Geometric graph neural networks (Geom-GNNs) with all-atom information have transformed atomistic simulations by serving as a general learnable geometric descriptors for downstream tasks including prediction of interatomic potential and molecular properties. However, common practices involve supervising Geom-GNNs on specific downstream tasks, which suffer from the lack of high-quality data and inaccurate labels leading to poor generalization and performance degradation on out-of-distribution (OOD) scenarios. In this work, we explored the possibility of using pre-trained Geom-GNNs as transferable and highly effective geometric descriptors for improved generalization. To explore their representation power, we studied the scaling behaviors of Geom-GNNs under self-supervised pre-training, supervised and unsupervised learning setups. We find that the expressive power of different architectures can differ on the pre-training task. Interestingly, Geom-GNNs do not follow the power-law scaling on the pre-training task, and universally lack predictable scaling behavior on the supervised tasks with quantum chemical labels important for screening and design of novel molecules. More importantly, we demonstrate how all-atom graph embedding can be organically combined with other neural architectures to enhance the expressive power. Meanwhile, the low-dimensional projection of the latent space shows excellent agreement with conventional geometrical descriptors.
geom2vec: pretrained GNNs as geometric featurizers for conformational dynamics
Sep 30, 2024
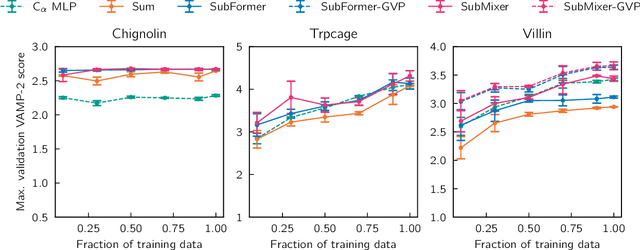
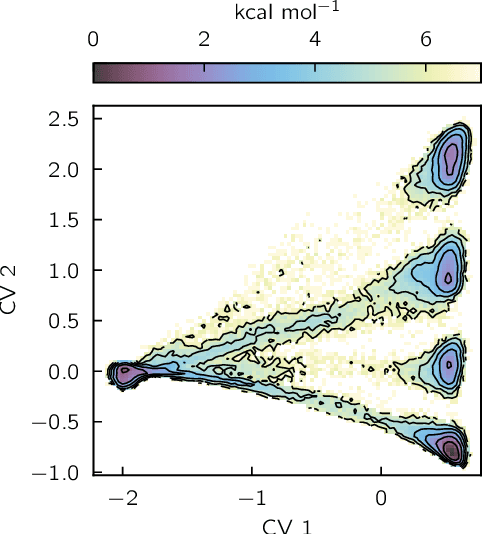
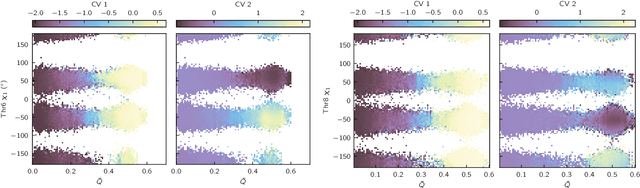
Identifying informative low-dimensional features that characterize dynamics in molecular simulations remains a challenge, often requiring extensive hand-tuning and system-specific knowledge. Here, we introduce geom2vec, in which pretrained graph neural networks (GNNs) are used as universal geometric featurizers. By pretraining equivariant GNNs on a large dataset of molecular conformations with a self-supervised denoising objective, we learn transferable structural representations that capture molecular geometric patterns without further fine-tuning. We show that the learned representations can be directly used to analyze trajectory data, thus eliminating the need for manual feature selection and improving robustness of the simulation analysis workflows. Importantly, by decoupling GNN training from training for downstream tasks, we enable analysis of larger molecular graphs with limited computational resources.
Technical Report: The Graph Spectral Token -- Enhancing Graph Transformers with Spectral Information
Apr 08, 2024Graph Transformers have emerged as a powerful alternative to Message-Passing Graph Neural Networks (MP-GNNs) to address limitations such as over-squashing of information exchange. However, incorporating graph inductive bias into transformer architectures remains a significant challenge. In this report, we propose the Graph Spectral Token, a novel approach to directly encode graph spectral information, which captures the global structure of the graph, into the transformer architecture. By parameterizing the auxiliary [CLS] token and leaving other tokens representing graph nodes, our method seamlessly integrates spectral information into the learning process. We benchmark the effectiveness of our approach by enhancing two existing graph transformers, GraphTrans and SubFormer. The improved GraphTrans, dubbed GraphTrans-Spec, achieves over 10% improvements on large graph benchmark datasets while maintaining efficiency comparable to MP-GNNs. SubFormer-Spec demonstrates strong performance across various datasets.
Transformers are efficient hierarchical chemical graph learners
Oct 02, 2023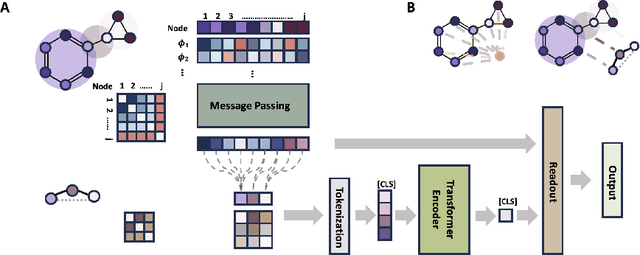

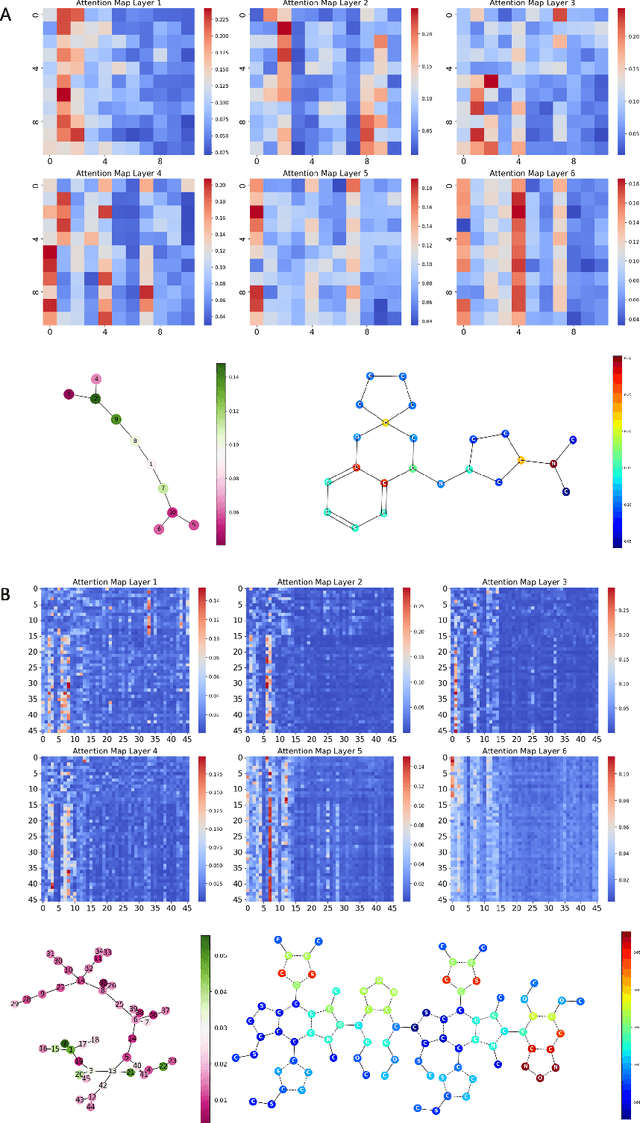
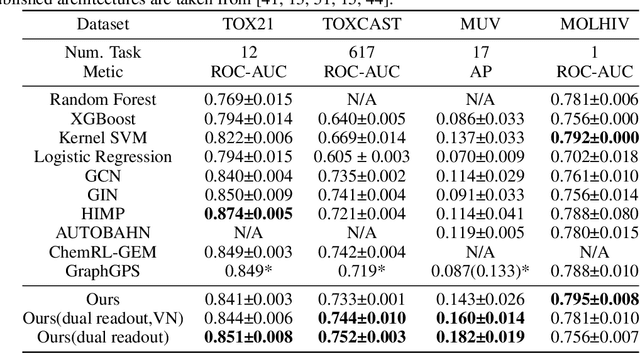
Transformers, adapted from natural language processing, are emerging as a leading approach for graph representation learning. Contemporary graph transformers often treat nodes or edges as separate tokens. This approach leads to computational challenges for even moderately-sized graphs due to the quadratic scaling of self-attention complexity with token count. In this paper, we introduce SubFormer, a graph transformer that operates on subgraphs that aggregate information by a message-passing mechanism. This approach reduces the number of tokens and enhances learning long-range interactions. We demonstrate SubFormer on benchmarks for predicting molecular properties from chemical structures and show that it is competitive with state-of-the-art graph transformers at a fraction of the computational cost, with training times on the order of minutes on a consumer-grade graphics card. We interpret the attention weights in terms of chemical structures. We show that SubFormer exhibits limited over-smoothing and avoids over-squashing, which is prevalent in traditional graph neural networks.
xxMD: Benchmarking Neural Force Fields Using Extended Dynamics beyond Equilibrium
Aug 30, 2023Neural force fields (NFFs) have gained prominence in computational chemistry as surrogate models, superseding quantum-chemistry calculations in ab initio molecular dynamics. The prevalent benchmark for NFFs has been the MD17 dataset and its subsequent extension. These datasets predominantly comprise geometries from the equilibrium region of the ground electronic state potential energy surface, sampling from direct adiabatic dynamics. However, many chemical reactions entail significant molecular deformations, notably bond breaking. We demonstrate the constrained distribution of internal coordinates and energies in the MD17 datasets, underscoring their inadequacy for representing systems undergoing chemical reactions. Addressing this sampling limitation, we introduce the xxMD (Extended Excited-state Molecular Dynamics) dataset, derived from non-adiabatic dynamics. This dataset encompasses energies and forces ascertained from both multireference wave function theory and density functional theory. Furthermore, its nuclear configuration spaces authentically depict chemical reactions, making xxMD a more chemically relevant dataset. Our re-assessment of equivariant models on the xxMD datasets reveals notably higher mean absolute errors than those reported for MD17 and its variants. This observation underscores the challenges faced in crafting a generalizable NFF model with extrapolation capability. Our proposed xxMD-CASSCF and xxMD-DFT datasets are available at https://github.com/zpengmei/xxMD.