 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edgegeom2vec: pretrained GNNs as geometric featurizers for conformational dynamics
Paper and Code
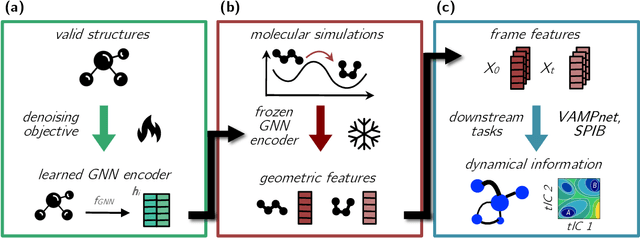
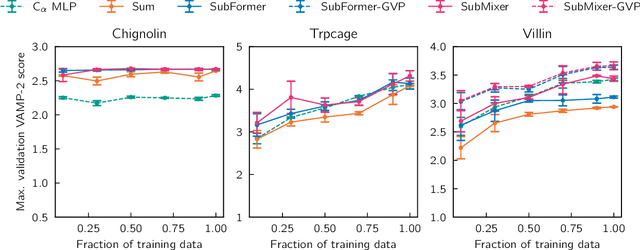
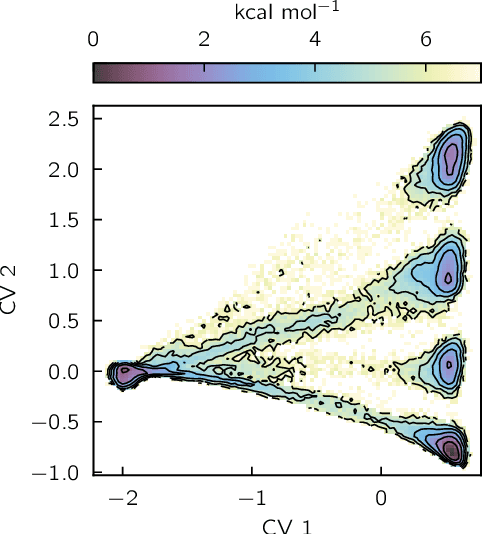
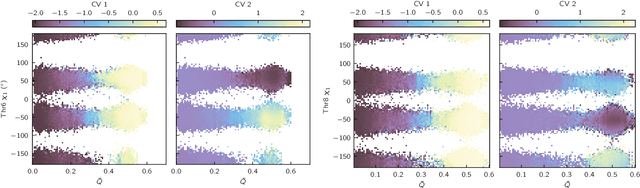
Identifying informative low-dimensional features that characterize dynamics in molecular simulations remains a challenge, often requiring extensive hand-tuning and system-specific knowledge. Here, we introduce geom2vec, in which pretrained graph neural networks (GNNs) are used as universal geometric featurizers. By pretraining equivariant GNNs on a large dataset of molecular conformations with a self-supervised denoising objective, we learn transferable structural representations that capture molecular geometric patterns without further fine-tuning. We show that the learned representations can be directly used to analyze trajectory data, thus eliminating the need for manual feature selection and improving robustness of the simulation analysis workflows. Importantly, by decoupling GNN training from training for downstream tasks, we enable analysis of larger molecular graphs with limited computational resources.