 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edgegeom2vec: pretrained GNNs as geometric featurizers for conformational dynamics
Sep 30, 2024
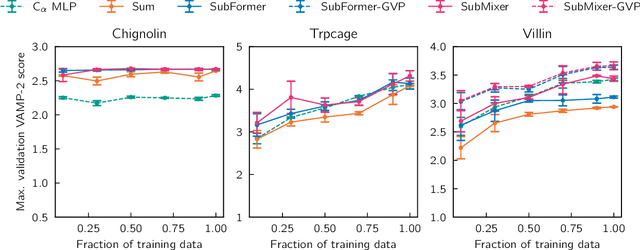
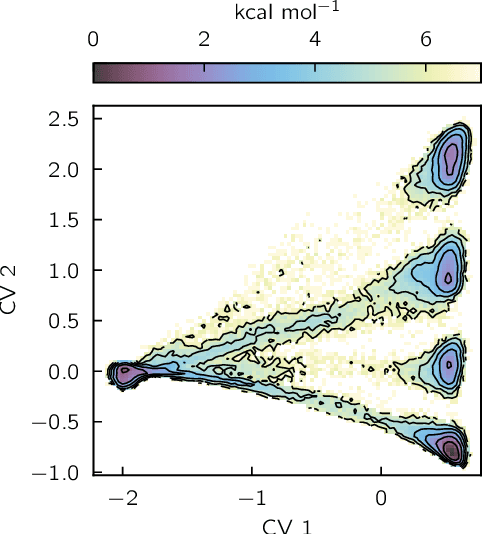
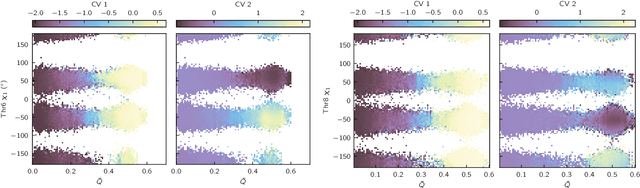
Identifying informative low-dimensional features that characterize dynamics in molecular simulations remains a challenge, often requiring extensive hand-tuning and system-specific knowledge. Here, we introduce geom2vec, in which pretrained graph neural networks (GNNs) are used as universal geometric featurizers. By pretraining equivariant GNNs on a large dataset of molecular conformations with a self-supervised denoising objective, we learn transferable structural representations that capture molecular geometric patterns without further fine-tuning. We show that the learned representations can be directly used to analyze trajectory data, thus eliminating the need for manual feature selection and improving robustness of the simulation analysis workflows. Importantly, by decoupling GNN training from training for downstream tasks, we enable analysis of larger molecular graphs with limited computational resources.
Inexact iterative numerical linear algebra for neural network-based spectral estimation and rare-event prediction
Mar 22, 2023Understanding dynamics in complex systems is challenging because there are many degrees of freedom, and those that are most important for describing events of interest are often not obvious. The leading eigenfunctions of the transition operator are useful for visualization, and they can provide an efficient basis for computing statistics such as the likelihood and average time of events (predictions). Here we develop inexact iterative linear algebra methods for computing these eigenfunctions (spectral estimation) and making predictions from a data set of short trajectories sampled at finite intervals. We demonstrate the methods on a low-dimensional model that facilitates visualization and a high-dimensional model of a biomolecular system. Implications for the prediction problem in reinforcement learning are discussed.