 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQM-ToT: A Medical Tree of Thoughts Reasoning Framework for Quantized Model
Apr 13, 2025Large language models (LLMs) face significant challenges in specialized biomedical tasks due to the inherent complexity of medical reasoning and the sensitive nature of clinical data. Existing LLMs often struggle with intricate medical terminology and the need for accurate clinical insights, leading to performance reduction when quantized for resource-constrained deployment. To address these issues, we propose Quantized Medical Tree of Thought (QM-ToT), a path-based reasoning framework. QM-ToT leverages a Tree of Thought (ToT) reasoning approach to decompose complex medical problems into manageable subtasks, coupled with evaluator assessment layers. This framework facilitates substantial performance improvements in INT4-quantized models on the challenging MedQAUSMLE dataset. Specifically, we demonstrate a remarkable accuracy increase from 34% to 50% for the LLaMA2-70b model and from 58.77% to 69.49% for LLaMA-3.1-8b. Besides, we also proposed an effect data distillation method based on ToT. Compared to the traditional distillation method, we achieved an improvement of 86. 27% while using only 3.9% of the data.This work, for the first time, showcases the potential of ToT to significantly enhance performance on complex biomedical tasks, establishing a crucial foundation for future advances in deploying high-performing quantized LLM in resource-limited medical settings.
Multimodal 3D Genome Pre-training
Apr 12, 2025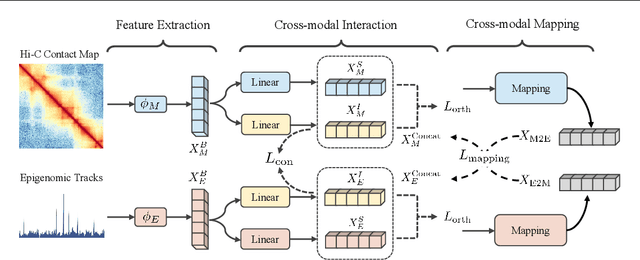
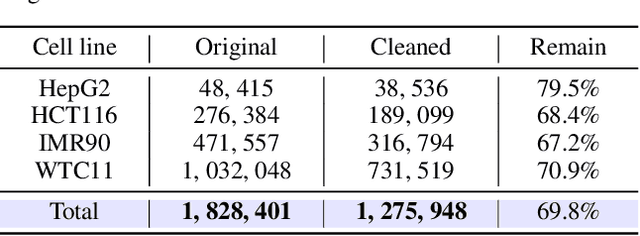
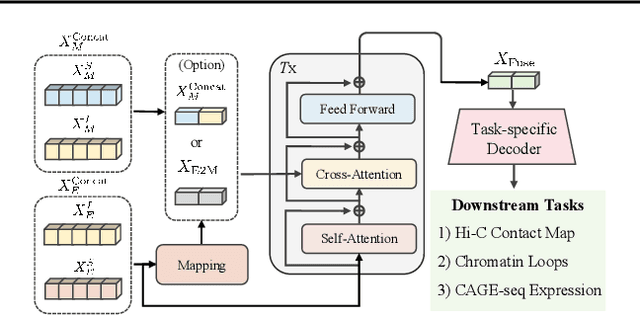
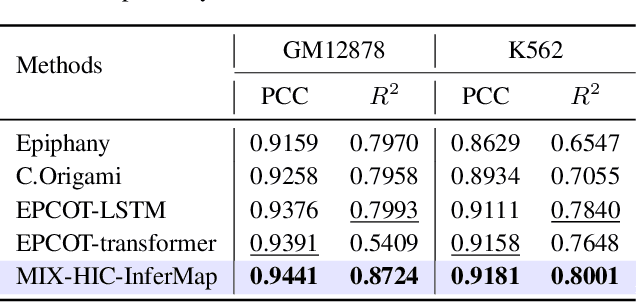
Deep learning techniques have driven significant progress in various analytical tasks within 3D genomics in computational biology. However, a holistic understanding of 3D genomics knowledge remains underexplored. Here, we propose MIX-HIC, the first multimodal foundation model of 3D genome that integrates both 3D genome structure and epigenomic tracks, which obtains unified and comprehensive semantics. For accurate heterogeneous semantic fusion, we design the cross-modal interaction and mapping blocks for robust unified representation, yielding the accurate aggregation of 3D genome knowledge. Besides, we introduce the first large-scale dataset comprising over 1 million pairwise samples of Hi-C contact maps and epigenomic tracks for high-quality pre-training, enabling the exploration of functional implications in 3D genomics. Extensive experiments show that MIX-HIC can significantly surpass existing state-of-the-art methods in diverse downstream tasks. This work provides a valuable resource for advancing 3D genomics research.
scBIT: Integrating Single-cell Transcriptomic Data into fMRI-based Prediction for Alzheimer's Disease Diagnosis
Feb 04, 2025



Functional MRI (fMRI) and single-cell transcriptomics are pivotal in Alzheimer's disease (AD) research, each providing unique insights into neural function and molecular mechanisms. However, integrating these complementary modalities remains largely unexplored. Here, we introduce scBIT, a novel method for enhancing AD prediction by combining fMRI with single-nucleus RNA (snRNA). scBIT leverages snRNA as an auxiliary modality, significantly improving fMRI-based prediction models and providing comprehensive interpretability. It employs a sampling strategy to segment snRNA data into cell-type-specific gene networks and utilizes a self-explainable graph neural network to extract critical subgraphs. Additionally, we use demographic and genetic similarities to pair snRNA and fMRI data across individuals, enabling robust cross-modal learning. Extensive experiments validate scBIT's effectiveness in revealing intricate brain region-gene associations and enhancing diagnostic prediction accuracy. By advancing brain imaging transcriptomics to the single-cell level, scBIT sheds new light on biomarker discovery in AD research. Experimental results show that incorporating snRNA data into the scBIT model significantly boosts accuracy, improving binary classification by 3.39% and five-class classification by 26.59%. The codes were implemented in Python and have been released on GitHub (https://github.com/77YQ77/scBIT) and Zenodo (https://zenodo.org/records/11599030) with detailed instructions.
Graph Structure Learning for Tumor Microenvironment with Cell Type Annotation from non-spatial scRNA-seq data
Feb 04, 2025The exploration of cellular heterogeneity within the tumor microenvironment (TME) via single-cell RNA sequencing (scRNA-seq) is essential for understanding cancer progression and response to therapy. Current scRNA-seq approaches, however, lack spatial context and rely on incomplete datasets of ligand-receptor interactions (LRIs), limiting accurate cell type annotation and cell-cell communication (CCC) inference. This study addresses these challenges using a novel graph neural network (GNN) model that enhances cell type prediction and cell interaction analysis. Our study utilized a dataset consisting of 49,020 cells from 19 patients across three cancer types: Leukemia, Breast Invasive Carcinoma, and Colorectal Cancer. The proposed scGSL model demonstrated robust performance, achieving an average accuracy of 84.83%, precision of 86.23%, recall of 81.51%, and an F1 score of 80.92% across all datasets. These metrics represent a significant enhancement over existing methods, which typically exhibit lower performance metrics. Additionally, by reviewing existing literature on gene interactions within the TME, the scGSL model proves to robustly identify biologically meaningful gene interactions in an unsupervised manner, validated by significant expression differences in key gene pairs across various cancers. The source code and data used in this paper can be found in https://github.com/LiYuechao1998/scGSL.
scGSDR: Harnessing Gene Semantics for Single-Cell Pharmacological Profiling
Feb 02, 2025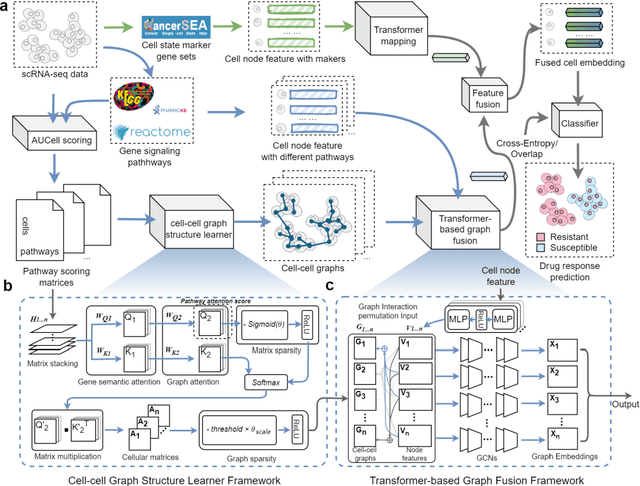



The rise of single-cell sequencing technologies has revolutionized the exploration of drug resistance, revealing the crucial role of cellular heterogeneity in advancing precision medicine. By building computational models from existing single-cell drug response data, we can rapidly annotate cellular responses to drugs in subsequent trials. To this end, we developed scGSDR, a model that integrates two computational pipelines grounded in the knowledge of cellular states and gene signaling pathways, both essential for understanding biological gene semantics. scGSDR enhances predictive performance by incorporating gene semantics and employs an interpretability module to identify key pathways contributing to drug resistance phenotypes. Our extensive validation, which included 16 experiments covering 11 drugs, demonstrates scGSDR's superior predictive accuracy, when trained with either bulk-seq or scRNA-seq data, achieving high AUROC, AUPR, and F1 Scores. The model's application has extended from single-drug predictions to scenarios involving drug combinations. Leveraging pathways of known drug target genes, we found that scGSDR's cell-pathway attention scores are biologically interpretable, which helped us identify other potential drug-related genes. Literature review of top-ranking genes in our predictions such as BCL2, CCND1, the AKT family, and PIK3CA for PLX4720; and ICAM1, VCAM1, NFKB1, NFKBIA, and RAC1 for Paclitaxel confirmed their relevance. In conclusion, scGSDR, by incorporating gene semantics, enhances predictive modeling of cellular responses to diverse drugs, proving invaluable for scenarios involving both single drug and combination therapies and effectively identifying key resistance-related pathways, thus advancing precision medicine and targeted therapy development.