 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe Power of 10: New Rules for the Digital World
Jan 07, 2026As artificial intelligence rapidly advances, society is increasingly captivated by promises of superhuman machines and seamless digital futures. Yet these visions often obscure mounting social, ethical, and psychological concerns tied to pervasive digital technologies - from surveillance to mental health crises. This article argues that a guiding ethos is urgently needed to navigate these transformations. Inspired by the lasting influence of the biblical Ten Commandments, a European interdisciplinary group has proposed "Ten Rules for the Digital World" - a novel ethical framework to help individuals and societies make prudent, human-centered decisions in the age of "supercharged" technology.
Predict Patient Self-reported Race from Skin Histological Images
Jul 30, 2025Artificial Intelligence (AI) has demonstrated success in computational pathology (CPath) for disease detection, biomarker classification, and prognosis prediction. However, its potential to learn unintended demographic biases, particularly those related to social determinants of health, remains understudied. This study investigates whether deep learning models can predict self-reported race from digitized dermatopathology slides and identifies potential morphological shortcuts. Using a multisite dataset with a racially diverse population, we apply an attention-based mechanism to uncover race-associated morphological features. After evaluating three dataset curation strategies to control for confounding factors, the final experiment showed that White and Black demographic groups retained high prediction performance (AUC: 0.799, 0.762), while overall performance dropped to 0.663. Attention analysis revealed the epidermis as a key predictive feature, with significant performance declines when these regions were removed. These findings highlight the need for careful data curation and bias mitigation to ensure equitable AI deployment in pathology. Code available at: https://github.com/sinai-computational-pathology/CPath_SAIF.
Screen Them All: High-Throughput Pan-Cancer Genetic and Phenotypic Biomarker Screening from H&E Whole Slide Images
Aug 20, 2024Many molecular alterations serve as clinically prognostic or therapy-predictive biomarkers, typically detected using single or multi-gene molecular assays. However, these assays are expensive, tissue destructive and often take weeks to complete. Using AI on routine H&E WSIs offers a fast and economical approach to screen for multiple molecular biomarkers. We present a high-throughput AI-based system leveraging Virchow2, a foundation model pre-trained on 3 million slides, to interrogate genomic features previously determined by an next-generation sequencing (NGS) assay, using 47,960 scanned hematoxylin and eosin (H&E) whole slide images (WSIs) from 38,984 cancer patients. Unlike traditional methods that train individual models for each biomarker or cancer type, our system employs a unified model to simultaneously predict a wide range of clinically relevant molecular biomarkers across cancer types. By training the network to replicate the MSK-IMPACT targeted biomarker panel of 505 genes, it identified 80 high performing biomarkers with a mean AU-ROC of 0.89 in 15 most common cancer types. In addition, 40 biomarkers demonstrated strong associations with specific cancer histologic subtypes. Furthermore, 58 biomarkers were associated with targets frequently assayed clinically for therapy selection and response prediction. The model can also predict the activity of five canonical signaling pathways, identify defects in DNA repair mechanisms, and predict genomic instability measured by tumor mutation burden, microsatellite instability (MSI), and chromosomal instability (CIN). The proposed model can offer potential to guide therapy selection, improve treatment efficacy, accelerate patient screening for clinical trials and provoke the interrogation of new therapeutic targets.
Virchow2: Scaling Self-Supervised Mixed Magnification Models in Pathology
Aug 14, 2024Foundation models are rapidly being developed for computational pathology applications. However, it remains an open question which factors are most important for downstream performance with data scale and diversity, model size, and training algorithm all playing a role. In this work, we propose algorithmic modifications, tailored for pathology, and we present the result of scaling both data and model size, surpassing previous studies in both dimensions. We introduce two new models: Virchow2, a 632 million parameter vision transformer, and Virchow2G, a 1.9 billion parameter vision transformer, each trained with 3.1 million histopathology whole slide images, with diverse tissues, originating institutions, and stains. We achieve state of the art performance on 12 tile-level tasks, as compared to the top performing competing models. Our results suggest that data diversity and domain-specific methods can outperform models that only scale in the number of parameters, but, on average, performance benefits from the combination of domain-specific methods, data scale, and model scale.
Virchow 2: Scaling Self-Supervised Mixed Magnification Models in Pathology
Aug 01, 2024Foundation models are rapidly being developed for computational pathology applications. However, it remains an open question which factors are most important for downstream performance with data scale and diversity, model size, and training algorithm all playing a role. In this work, we present the result of scaling both data and model size, surpassing previous studies in both dimensions, and introduce two new models: Virchow 2, a 632M parameter vision transformer, and Virchow 2G, a 1.85B parameter vision transformer, each trained with 3.1M histopathology whole slide images. To support this scale, we propose domain-inspired adaptations to the DINOv2 training algorithm, which is quickly becoming the default method in self-supervised learning for computational pathology. We achieve state of the art performance on twelve tile-level tasks, as compared to the top performing competing models. Our results suggest that data diversity and domain-specific training can outperform models that only scale in the number of parameters, but, on average, performance benefits from domain-tailoring, data scale, and model scale.
Multi-Organ Cancer Classification and Survival Analysis
Dec 02, 2016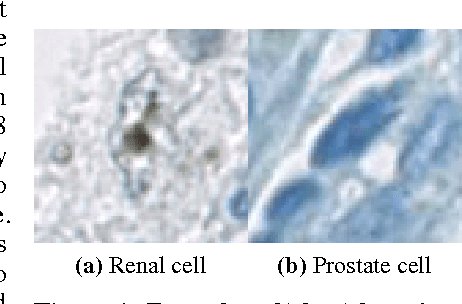



Accurate and robust cell nuclei classification is the cornerstone for a wider range of tasks in digital and Computational Pathology. However, most machine learning systems require extensive labeling from expert pathologists for each individual problem at hand, with no or limited abilities for knowledge transfer between datasets and organ sites. In this paper we implement and evaluate a variety of deep neural network models and model ensembles for nuclei classification in renal cell cancer (RCC) and prostate cancer (PCa). We propose a convolutional neural network system based on residual learning which significantly improves over the state-of-the-art in cell nuclei classification. Finally, we show that the combination of tissue types during training increases not only classification accuracy but also overall survival analysis.
Understanding Neural Networks Through Deep Visualization
Jun 22, 2015


Recent years have produced great advances in training large, deep neural networks (DNNs), including notable successes in training convolutional neural networks (convnets) to recognize natural images. However, our understanding of how these models work, especially what computations they perform at intermediate layers, has lagged behind. Progress in the field will be further accelerated by the development of better tools for visualizing and interpreting neural nets. We introduce two such tools here. The first is a tool that visualizes the activations produced on each layer of a trained convnet as it processes an image or video (e.g. a live webcam stream). We have found that looking at live activations that change in response to user input helps build valuable intuitions about how convnets work. The second tool enables visualizing features at each layer of a DNN via regularized optimization in image space. Because previous versions of this idea produced less recognizable images, here we introduce several new regularization methods that combine to produce qualitatively clearer, more interpretable visualizations. Both tools are open source and work on a pre-trained convnet with minimal setup.