 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSingle GPU Task Adaptation of Pathology Foundation Models for Whole Slide Image Analysis
Jun 05, 2025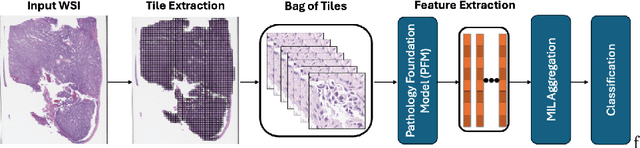
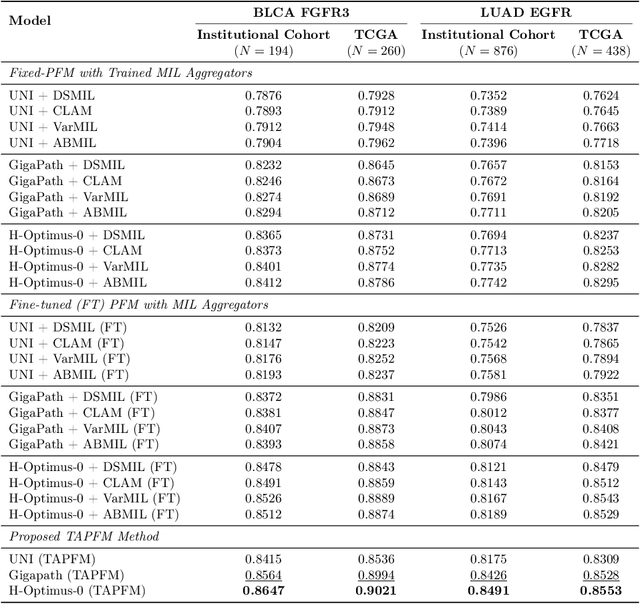
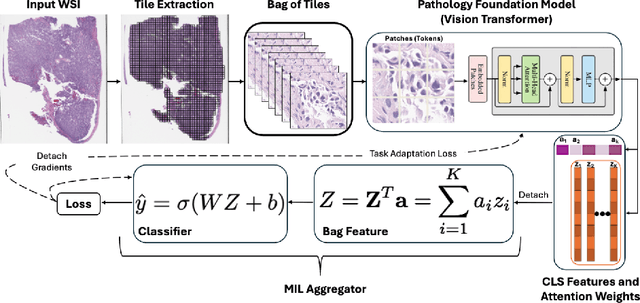
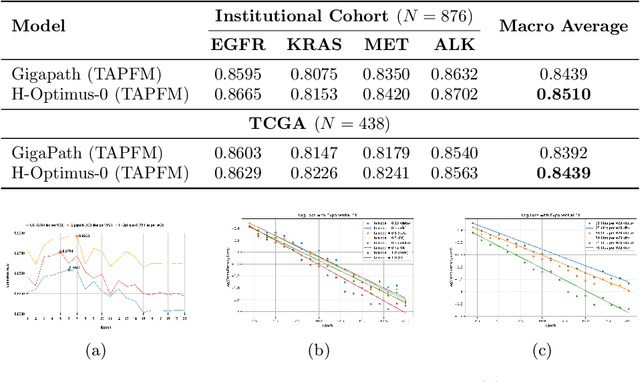
Pathology foundation models (PFMs) have emerged as powerful tools for analyzing whole slide images (WSIs). However, adapting these pretrained PFMs for specific clinical tasks presents considerable challenges, primarily due to the availability of only weak (WSI-level) labels for gigapixel images, necessitating multiple instance learning (MIL) paradigm for effective WSI analysis. This paper proposes a novel approach for single-GPU \textbf{T}ask \textbf{A}daptation of \textbf{PFM}s (TAPFM) that uses vision transformer (\vit) attention for MIL aggregation while optimizing both for feature representations and attention weights. The proposed approach maintains separate computational graphs for MIL aggregator and the PFM to create stable training dynamics that align with downstream task objectives during end-to-end adaptation. Evaluated on mutation prediction tasks for bladder cancer and lung adenocarcinoma across institutional and TCGA cohorts, TAPFM consistently outperforms conventional approaches, with H-Optimus-0 (TAPFM) outperforming the benchmarks. TAPFM effectively handles multi-label classification of actionable mutations as well. Thus, TAPFM makes adaptation of powerful pre-trained PFMs practical on standard hardware for various clinical applications.
Screen Them All: High-Throughput Pan-Cancer Genetic and Phenotypic Biomarker Screening from H&E Whole Slide Images
Aug 20, 2024Many molecular alterations serve as clinically prognostic or therapy-predictive biomarkers, typically detected using single or multi-gene molecular assays. However, these assays are expensive, tissue destructive and often take weeks to complete. Using AI on routine H&E WSIs offers a fast and economical approach to screen for multiple molecular biomarkers. We present a high-throughput AI-based system leveraging Virchow2, a foundation model pre-trained on 3 million slides, to interrogate genomic features previously determined by an next-generation sequencing (NGS) assay, using 47,960 scanned hematoxylin and eosin (H&E) whole slide images (WSIs) from 38,984 cancer patients. Unlike traditional methods that train individual models for each biomarker or cancer type, our system employs a unified model to simultaneously predict a wide range of clinically relevant molecular biomarkers across cancer types. By training the network to replicate the MSK-IMPACT targeted biomarker panel of 505 genes, it identified 80 high performing biomarkers with a mean AU-ROC of 0.89 in 15 most common cancer types. In addition, 40 biomarkers demonstrated strong associations with specific cancer histologic subtypes. Furthermore, 58 biomarkers were associated with targets frequently assayed clinically for therapy selection and response prediction. The model can also predict the activity of five canonical signaling pathways, identify defects in DNA repair mechanisms, and predict genomic instability measured by tumor mutation burden, microsatellite instability (MSI), and chromosomal instability (CIN). The proposed model can offer potential to guide therapy selection, improve treatment efficacy, accelerate patient screening for clinical trials and provoke the interrogation of new therapeutic targets.
A Clinical Benchmark of Public Self-Supervised Pathology Foundation Models
Jul 11, 2024



The use of self-supervised learning (SSL) to train pathology foundation models has increased substantially in the past few years. Notably, several models trained on large quantities of clinical data have been made publicly available in recent months. This will significantly enhance scientific research in computational pathology and help bridge the gap between research and clinical deployment. With the increase in availability of public foundation models of different sizes, trained using different algorithms on different datasets, it becomes important to establish a benchmark to compare the performance of such models on a variety of clinically relevant tasks spanning multiple organs and diseases. In this work, we present a collection of pathology datasets comprising clinical slides associated with clinically relevant endpoints including cancer diagnoses and a variety of biomarkers generated during standard hospital operation from two medical centers. We leverage these datasets to systematically assess the performance of public pathology foundation models and provide insights into best practices for training new foundation models and selecting appropriate pretrained models.
Benchmarking Embedding Aggregation Methods in Computational Pathology: A Clinical Data Perspective
Jul 10, 2024


Recent advances in artificial intelligence (AI), in particular self-supervised learning of foundation models (FMs), are revolutionizing medical imaging and computational pathology (CPath). A constant challenge in the analysis of digital Whole Slide Images (WSIs) is the problem of aggregating tens of thousands of tile-level image embeddings to a slide-level representation. Due to the prevalent use of datasets created for genomic research, such as TCGA, for method development, the performance of these techniques on diagnostic slides from clinical practice has been inadequately explored. This study conducts a thorough benchmarking analysis of ten slide-level aggregation techniques across nine clinically relevant tasks, including diagnostic assessment, biomarker classification, and outcome prediction. The results yield following key insights: (1) Embeddings derived from domain-specific (histological images) FMs outperform those from generic ImageNet-based models across aggregation methods. (2) Spatial-aware aggregators enhance the performance significantly when using ImageNet pre-trained models but not when using FMs. (3) No single model excels in all tasks and spatially-aware models do not show general superiority as it would be expected. These findings underscore the need for more adaptable and universally applicable aggregation techniques, guiding future research towards tools that better meet the evolving needs of clinical-AI in pathology. The code used in this work is available at \url{https://github.com/fuchs-lab-public/CPath_SABenchmark}.
Beyond Multiple Instance Learning: Full Resolution All-In-Memory End-To-End Pathology Slide Modeling
Mar 07, 2024


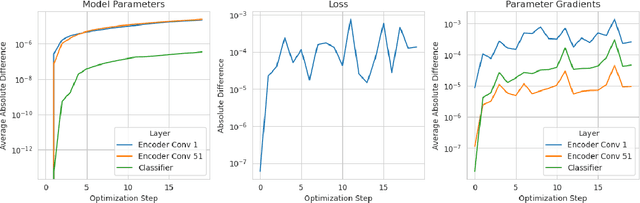
Artificial Intelligence (AI) has great potential to improve health outcomes by training systems on vast digitized clinical datasets. Computational Pathology, with its massive amounts of microscopy image data and impact on diagnostics and biomarkers, is at the forefront of this development. Gigapixel pathology slides pose a unique challenge due to their enormous size and are usually divided into tens of thousands of smaller tiles for analysis. This results in a discontinuity in the machine learning process by separating the training of tile-level encoders from slide-level aggregators and the need to adopt weakly supervised learning strategies. Training models from entire pathology slides end-to-end has been largely unexplored due to its computational challenges. To overcome this problem, we propose a novel approach to jointly train both a tile encoder and a slide-aggregator fully in memory and end-to-end at high-resolution, bridging the gap between input and slide-level supervision. While more computationally expensive, detailed quantitative validation shows promise for large-scale pre-training of pathology foundation models.
Computational Pathology at Health System Scale -- Self-Supervised Foundation Models from Three Billion Images
Oct 10, 2023Recent breakthroughs in self-supervised learning have enabled the use of large unlabeled datasets to train visual foundation models that can generalize to a variety of downstream tasks. While this training paradigm is well suited for the medical domain where annotations are scarce, large-scale pre-training in the medical domain, and in particular pathology, has not been extensively studied. Previous work in self-supervised learning in pathology has leveraged smaller datasets for both pre-training and evaluating downstream performance. The aim of this project is to train the largest academic foundation model and benchmark the most prominent self-supervised learning algorithms by pre-training and evaluating downstream performance on large clinical pathology datasets. We collected the largest pathology dataset to date, consisting of over 3 billion images from over 423 thousand microscopy slides. We compared pre-training of visual transformer models using the masked autoencoder (MAE) and DINO algorithms. We evaluated performance on six clinically relevant tasks from three anatomic sites and two institutions: breast cancer detection, inflammatory bowel disease detection, breast cancer estrogen receptor prediction, lung adenocarcinoma EGFR mutation prediction, and lung cancer immunotherapy response prediction. Our results demonstrate that pre-training on pathology data is beneficial for downstream performance compared to pre-training on natural images. Additionally, the DINO algorithm achieved better generalization performance across all tasks tested. The presented results signify a phase change in computational pathology research, paving the way into a new era of more performant models based on large-scale, parallel pre-training at the billion-image scale.
H&E-based Computational Biomarker Enables Universal EGFR Screening for Lung Adenocarcinoma
Jun 21, 2022
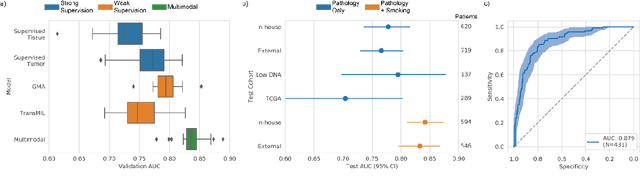
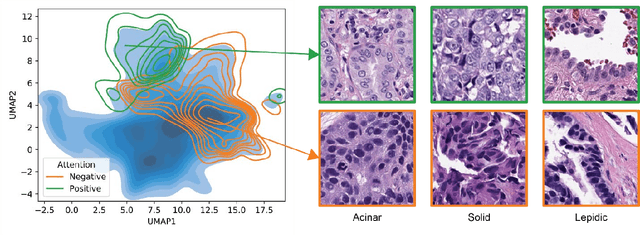
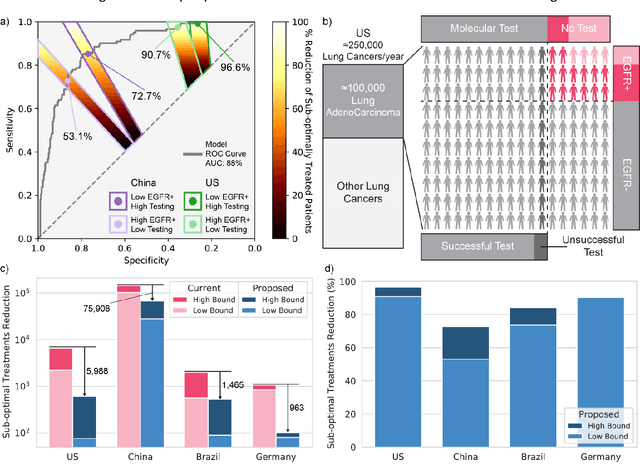
Lung cancer is the leading cause of cancer death worldwide, with lung adenocarcinoma being the most prevalent form of lung cancer. EGFR positive lung adenocarcinomas have been shown to have high response rates to TKI therapy, underlying the essential nature of molecular testing for lung cancers. Despite current guidelines consider testing necessary, a large portion of patients are not routinely profiled, resulting in millions of people not receiving the optimal treatment for their lung cancer. Sequencing is the gold standard for molecular testing of EGFR mutations, but it can take several weeks for results to come back, which is not ideal in a time constrained scenario. The development of alternative screening tools capable of detecting EGFR mutations quickly and cheaply while preserving tissue for sequencing could help reduce the amount of sub-optimally treated patients. We propose a multi-modal approach which integrates pathology images and clinical variables to predict EGFR mutational status achieving an AUC of 84% on the largest clinical cohort to date. Such a computational model could be deployed at large at little additional cost. Its clinical application could reduce the number of patients who receive sub-optimal treatments by 53.1% in China, and up to 96.6% in the US.