 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePRISM2: Unlocking Multi-Modal General Pathology AI with Clinical Dialogue
Jun 16, 2025Recent pathology foundation models can provide rich tile-level representations but fall short of delivering general-purpose clinical utility without further extensive model development. These models lack whole-slide image (WSI) understanding and are not trained with large-scale diagnostic data, limiting their performance on diverse downstream tasks. We introduce PRISM2, a multi-modal slide-level foundation model trained via clinical dialogue to enable scalable, generalizable pathology AI. PRISM2 is trained on nearly 700,000 specimens (2.3 million WSIs) paired with real-world clinical diagnostic reports in a two-stage process. In Stage 1, a vision-language model is trained using contrastive and captioning objectives to align whole slide embeddings with textual clinical diagnosis. In Stage 2, the language model is unfrozen to enable diagnostic conversation and extract more clinically meaningful representations from hidden states. PRISM2 achieves strong performance on diagnostic and biomarker prediction tasks, outperforming prior slide-level models including PRISM and TITAN. It also introduces a zero-shot yes/no classification approach that surpasses CLIP-style methods without prompt tuning or class enumeration. By aligning visual features with clinical reasoning, PRISM2 improves generalization on both data-rich and low-sample tasks, offering a scalable path forward for building general pathology AI agents capable of assisting diagnostic and prognostic decisions.
Screen Them All: High-Throughput Pan-Cancer Genetic and Phenotypic Biomarker Screening from H&E Whole Slide Images
Aug 20, 2024Many molecular alterations serve as clinically prognostic or therapy-predictive biomarkers, typically detected using single or multi-gene molecular assays. However, these assays are expensive, tissue destructive and often take weeks to complete. Using AI on routine H&E WSIs offers a fast and economical approach to screen for multiple molecular biomarkers. We present a high-throughput AI-based system leveraging Virchow2, a foundation model pre-trained on 3 million slides, to interrogate genomic features previously determined by an next-generation sequencing (NGS) assay, using 47,960 scanned hematoxylin and eosin (H&E) whole slide images (WSIs) from 38,984 cancer patients. Unlike traditional methods that train individual models for each biomarker or cancer type, our system employs a unified model to simultaneously predict a wide range of clinically relevant molecular biomarkers across cancer types. By training the network to replicate the MSK-IMPACT targeted biomarker panel of 505 genes, it identified 80 high performing biomarkers with a mean AU-ROC of 0.89 in 15 most common cancer types. In addition, 40 biomarkers demonstrated strong associations with specific cancer histologic subtypes. Furthermore, 58 biomarkers were associated with targets frequently assayed clinically for therapy selection and response prediction. The model can also predict the activity of five canonical signaling pathways, identify defects in DNA repair mechanisms, and predict genomic instability measured by tumor mutation burden, microsatellite instability (MSI), and chromosomal instability (CIN). The proposed model can offer potential to guide therapy selection, improve treatment efficacy, accelerate patient screening for clinical trials and provoke the interrogation of new therapeutic targets.
Virchow2: Scaling Self-Supervised Mixed Magnification Models in Pathology
Aug 14, 2024Foundation models are rapidly being developed for computational pathology applications. However, it remains an open question which factors are most important for downstream performance with data scale and diversity, model size, and training algorithm all playing a role. In this work, we propose algorithmic modifications, tailored for pathology, and we present the result of scaling both data and model size, surpassing previous studies in both dimensions. We introduce two new models: Virchow2, a 632 million parameter vision transformer, and Virchow2G, a 1.9 billion parameter vision transformer, each trained with 3.1 million histopathology whole slide images, with diverse tissues, originating institutions, and stains. We achieve state of the art performance on 12 tile-level tasks, as compared to the top performing competing models. Our results suggest that data diversity and domain-specific methods can outperform models that only scale in the number of parameters, but, on average, performance benefits from the combination of domain-specific methods, data scale, and model scale.
PRISM: A Multi-Modal Generative Foundation Model for Slide-Level Histopathology
May 16, 2024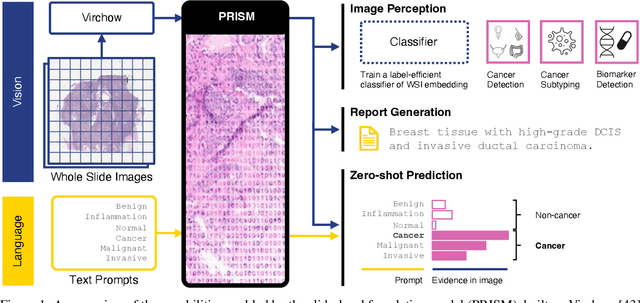
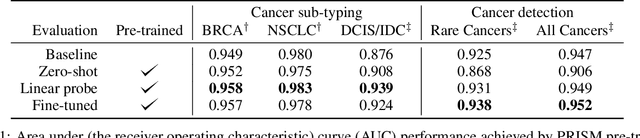
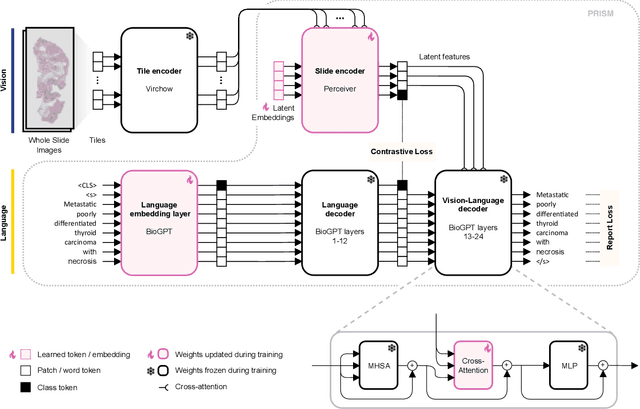
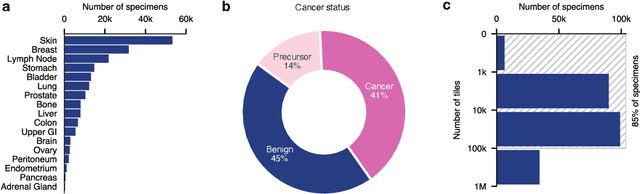
Foundation models in computational pathology promise to unlock the development of new clinical decision support systems and models for precision medicine. However, there is a mismatch between most clinical analysis, which is defined at the level of one or more whole slide images, and foundation models to date, which process the thousands of image tiles contained in a whole slide image separately. The requirement to train a network to aggregate information across a large number of tiles in multiple whole slide images limits these models' impact. In this work, we present a slide-level foundation model for H&E-stained histopathology, PRISM, that builds on Virchow tile embeddings and leverages clinical report text for pre-training. Using the tile embeddings, PRISM produces slide-level embeddings with the ability to generate clinical reports, resulting in several modes of use. Using text prompts, PRISM achieves zero-shot cancer detection and sub-typing performance approaching and surpassing that of a supervised aggregator model. Using the slide embeddings with linear classifiers, PRISM surpasses supervised aggregator models. Furthermore, we demonstrate that fine-tuning of the PRISM slide encoder yields label-efficient training for biomarker prediction, a task that typically suffers from low availability of training data; an aggregator initialized with PRISM and trained on as little as 10% of the training data can outperform a supervised baseline that uses all of the data.
Virchow: A Million-Slide Digital Pathology Foundation Model
Sep 21, 2023Computational pathology uses artificial intelligence to enable precision medicine and decision support systems through the analysis of whole slide images. It has the potential to revolutionize the diagnosis and treatment of cancer. However, a major challenge to this objective is that for many specific computational pathology tasks the amount of data is inadequate for development. To address this challenge, we created Virchow, a 632 million parameter deep neural network foundation model for computational pathology. Using self-supervised learning, Virchow is trained on 1.5 million hematoxylin and eosin stained whole slide images from diverse tissue groups, which is orders of magnitude more data than previous works. When evaluated on downstream tasks including tile-level pan-cancer detection and subtyping and slide-level biomarker prediction, Virchow outperforms state-of-the-art systems both on internal datasets drawn from the same population as the pretraining data as well as external public datasets. Virchow achieves 93% balanced accuracy for pancancer tile classification, and AUCs of 0.983 for colon microsatellite instability status prediction and 0.967 for breast CDH1 status prediction. The gains in performance highlight the importance of pretraining on massive pathology image datasets, suggesting pretraining on even larger datasets could continue improving performance for many high-impact applications where limited amounts of training data are available, such as drug outcome prediction.