 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDraft Less, Retrieve More: Hybrid Tree Construction for Speculative Decoding
May 19, 2026Speculative decoding (SD) accelerates large language model inference by leveraging a draft-then-verify paradigm. To maximize the acceptance rate, recent methods construct expansive draft trees, which unfortunately incur severe VRAM bandwidth and computational overheads that bottleneck end-to-end speedups. While dynamic-depth pruning can reduce this latency by removing marginal branches, it also discards potentially valid candidates, preventing the acceptance rate from reaching the upper bound of dense trees. In this paper, we identify a critical opportunity in resource allocation: the transition from dense to pruned drafting frees up significant computational budget. To break this Pareto tradeoff, we introduce Graft, a compensation framework that couples pruning and retrieval as mutually reinforcing operations. Pruning supplies sufficient budget for retrieval, while retrieval compensates for pruning-induced coverage loss and recovers accepted length. By employing a sequential `prune-then-graft' mechanism, Graft attaches highly predictive retrieved tokens into positions opened by pruning, filling the topological gaps with near-zero overhead. Graft is entirely training-free and lossless. Comprehensive evaluations show that Graft establishes a new Pareto frontier across practical deployment settings, including short-context generation, long-context generation, and large-scale models. On short-context benchmarks, it achieves up to 5.41$\times$ speedup and improves average speedup over EAGLE-3 by up to 21.8% on the large-scale Qwen3-235B. We also provide a preliminary exploration of applying Graft to the DFlash-style block drafting paradigm, offering initial evidence and insights for extending grafting beyond autoregressive draft trees.
A Multi-task Large Reasoning Model for Molecular Science
Mar 13, 2026Advancements in artificial intelligence for molecular science are necessitating a paradigm shift from purely data-driven predictions to knowledge-guided computational reasoning. Existing molecular models are predominantly proprietary, lacking general molecular intelligence and generalizability. This underscores the necessity for computational methods that can effectively integrate scientific logic with deep learning architectures. Here we introduce a multi-task large reasoning model designed to emulate the cognitive processes of molecular scientists through structured reasoning and reflection. Our approach incorporates multi-specialist modules to provide versatile molecular expertise and a chain-of-thought (CoT) framework enhanced by reinforcement learning infused with molecular knowledge, enabling structured and reflective reasoning. Systematic evaluations across 10 molecular tasks and 47 metrics demonstrate that our model achieves an average 50.3% improvement over the base architecture, outperforming over 20 state-of-the-art baselines, including ultra-large-parameter foundation models, despite using significantly fewer training data and computational resources. This validates that embedding explicit reasoning mechanisms enables high-efficiency learning, allowing smaller-scale models to surpass massive counterparts in both efficacy and interpretability. The practical utility of this computational framework was validated through a case study on the design of central nervous system (CNS) drug candidates, illustrating its capacity to bridge data-driven and knowledge-integrated approaches for intelligent molecular design.
GloPath: An Entity-Centric Foundation Model for Glomerular Lesion Assessment and Clinicopathological Insights
Mar 03, 2026Glomerular pathology is central to the diagnosis and prognosis of renal diseases, yet the heterogeneity of glomerular morphology and fine-grained lesion patterns remain challenging for current AI approaches. We present GloPath, an entity-centric foundation model trained on over one million glomeruli extracted from 14,049 renal biopsy specimens using multi-scale and multi-view self-supervised learning. GloPath addresses two major challenges in nephropathology: glomerular lesion assessment and clinicopathological insights discovery. For lesion assessment, GloPath was benchmarked across three independent cohorts on 52 tasks, including lesion recognition, grading, few-shot classification, and cross-modality diagnosis-outperforming state-of-the-art methods in 42 tasks (80.8%). In the large-scale real-world study, it achieved an ROC-AUC of 91.51% for lesion recognition, demonstrating strong robustness in routine clinical settings. For clinicopathological insights, GloPath systematically revealed statistically significant associations between glomerular morphological parameters and clinical indicators across 224 morphology-clinical variable pairs, demonstrating its capacity to connect tissue-level pathology with patient-level outcomes. Together, these results position GloPath as a scalable and interpretable platform for glomerular lesion assessment and clinicopathological discovery, representing a step toward clinically translatable AI in renal pathology.
Deep Learning in Single-Cell and Spatial Transcriptomics Data Analysis: Advances and Challenges from a Data Science Perspective
Dec 04, 2024



The development of single-cell and spatial transcriptomics has revolutionized our capacity to investigate cellular properties, functions, and interactions in both cellular and spatial contexts. However, the analysis of single-cell and spatial omics data remains challenging. First, single-cell sequencing data are high-dimensional and sparse, often contaminated by noise and uncertainty, obscuring the underlying biological signals. Second, these data often encompass multiple modalities, including gene expression, epigenetic modifications, and spatial locations. Integrating these diverse data modalities is crucial for enhancing prediction accuracy and biological interpretability. Third, while the scale of single-cell sequencing has expanded to millions of cells, high-quality annotated datasets are still limited. Fourth, the complex correlations of biological tissues make it difficult to accurately reconstruct cellular states and spatial contexts. Traditional feature engineering-based analysis methods struggle to deal with the various challenges presented by intricate biological networks. Deep learning has emerged as a powerful tool capable of handling high-dimensional complex data and automatically identifying meaningful patterns, offering significant promise in addressing these challenges. This review systematically analyzes these challenges and discusses related deep learning approaches. Moreover, we have curated 21 datasets from 9 benchmarks, encompassing 58 computational methods, and evaluated their performance on the respective modeling tasks. Finally, we highlight three areas for future development from a technical, dataset, and application perspective. This work will serve as a valuable resource for understanding how deep learning can be effectively utilized in single-cell and spatial transcriptomics analyses, while inspiring novel approaches to address emerging challenges.
BSM loss: A superior way in modeling aleatory uncertainty of fine_grained classification
Jun 09, 2022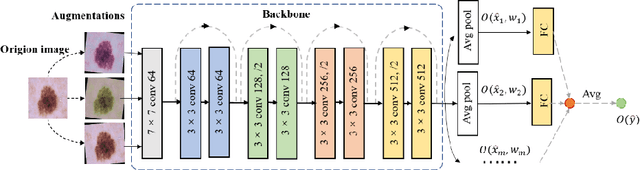
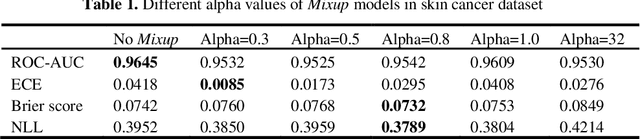
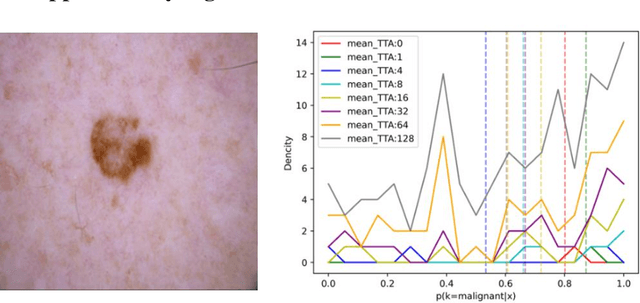
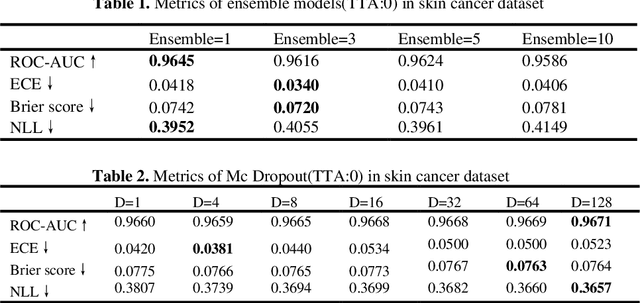
Artificial intelligence(AI)-assisted method had received much attention in the risk field such as disease diagnosis. Different from the classification of disease types, it is a fine-grained task to classify the medical images as benign or malignant. However, most research only focuses on improving the diagnostic accuracy and ignores the evaluation of model reliability, which limits its clinical application. For clinical practice, calibration presents major challenges in the low-data regime extremely for over-parametrized models and inherent noises. In particular, we discovered that modeling data-dependent uncertainty is more conducive to confidence calibrations. Compared with test-time augmentation(TTA), we proposed a modified Bootstrapping loss(BS loss) function with Mixup data augmentation strategy that can better calibrate predictive uncertainty and capture data distribution transformation without additional inference time. Our experiments indicated that BS loss with Mixup(BSM) model can halve the Expected Calibration Error(ECE) compared to standard data augmentation, deep ensemble and MC dropout. The correlation between uncertainty and similarity of in-domain data is up to -0.4428 under the BSM model. Additionally, the BSM model is able to perceive the semantic distance of out-of-domain data, demonstrating high potential in real-world clinical practice.
MIPR:Automatic Annotation of Medical Images with Pixel Rearrangement
Apr 22, 2022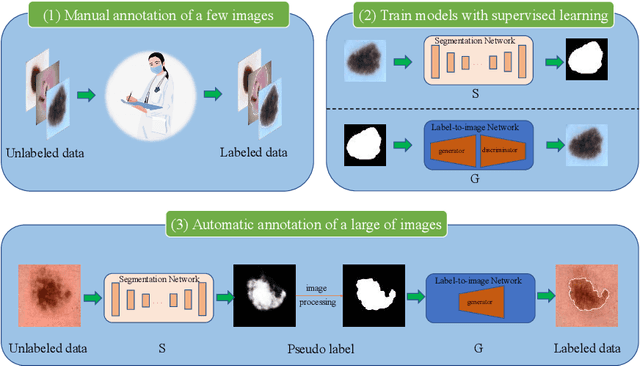
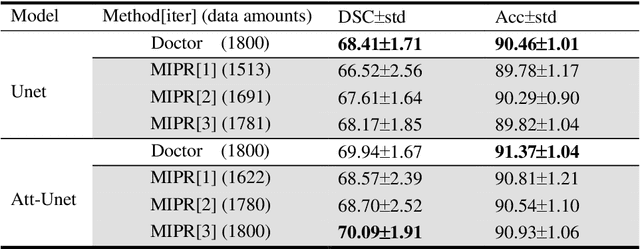
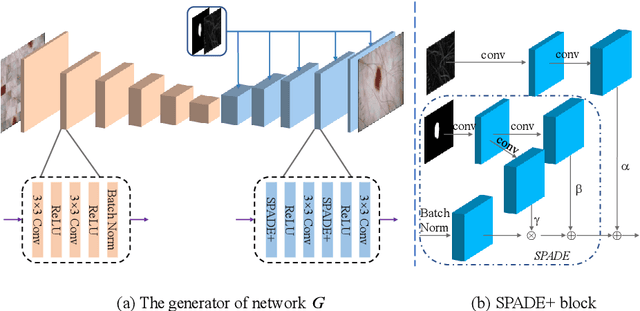

Most of the state-of-the-art semantic segmentation reported in recent years is based on fully supervised deep learning in the medical domain. How?ever, the high-quality annotated datasets require intense labor and domain knowledge, consuming enormous time and cost. Previous works that adopt semi?supervised and unsupervised learning are proposed to address the lack of anno?tated data through assisted training with unlabeled data and achieve good perfor?mance. Still, these methods can not directly get the image annotation as doctors do. In this paper, inspired by self-training of semi-supervised learning, we pro?pose a novel approach to solve the lack of annotated data from another angle, called medical image pixel rearrangement (short in MIPR). The MIPR combines image-editing and pseudo-label technology to obtain labeled data. As the number of iterations increases, the edited image is similar to the original image, and the labeled result is similar to the doctor annotation. Therefore, the MIPR is to get labeled pairs of data directly from amounts of unlabled data with pixel rearrange?ment, which is implemented with a designed conditional Generative Adversarial Networks and a segmentation network. Experiments on the ISIC18 show that the effect of the data annotated by our method for segmentation task is is equal to or even better than that of doctors annotations
AI assisted method for efficiently generating breast ultrasound screening reports
Jul 28, 2021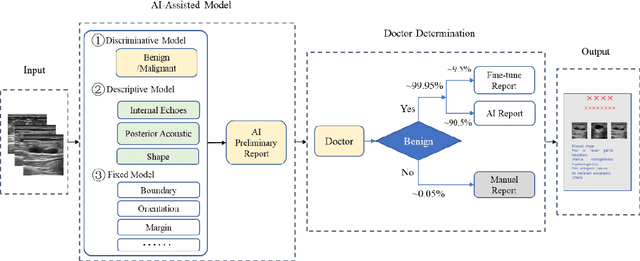

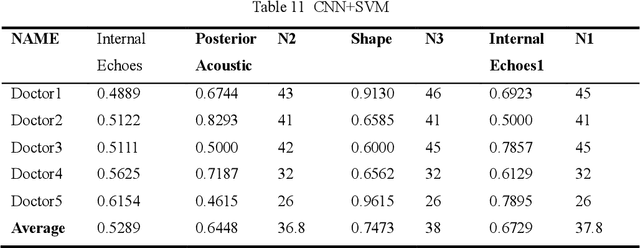
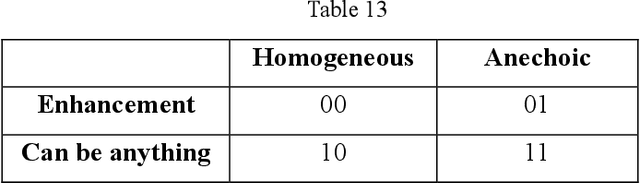
Ultrasound is the preferred choice for early screening of dense breast cancer. Clinically, doctors have to manually write the screening report which is time-consuming and laborious, and it is easy to miss and miswrite. Therefore, this paper proposes a method for efficiently generating personalized breast ultrasound screening preliminary reports by AI, especially for benign and normal cases which account for the majority. Doctors then make simple adjustments or corrections to quickly generate final reports. The proposed approach has been tested using a database of 1133 breast tumor instances. Experimental results indicate this pipeline improves doctors' work efficiency by up to 90%, which greatly reduces repetitive work.