 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEvery Step Evolves: Scaling Reinforcement Learning for Trillion-Scale Thinking Model
Oct 21, 2025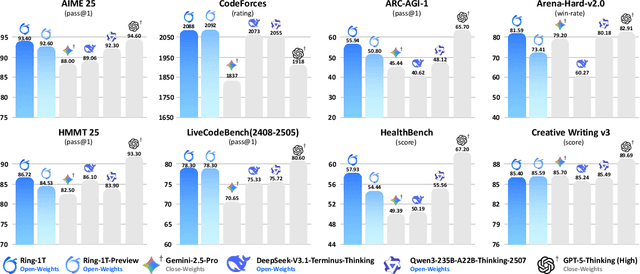
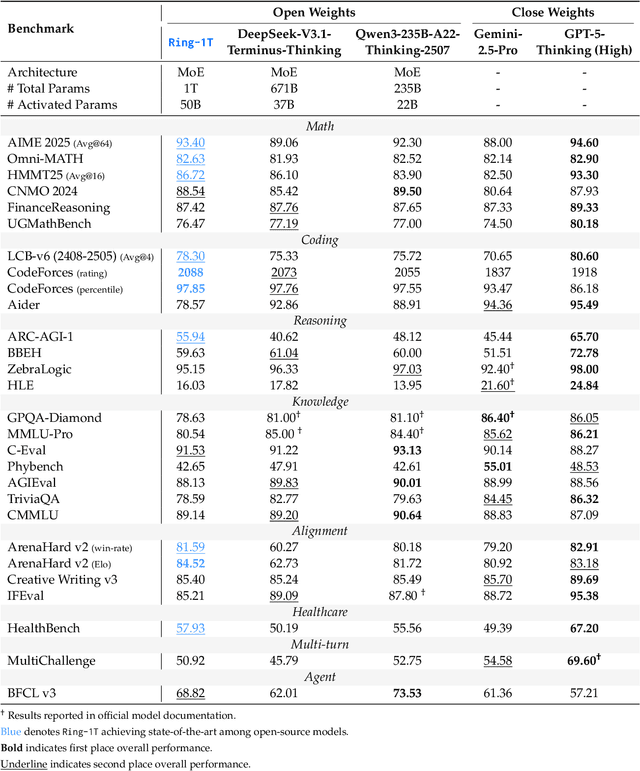
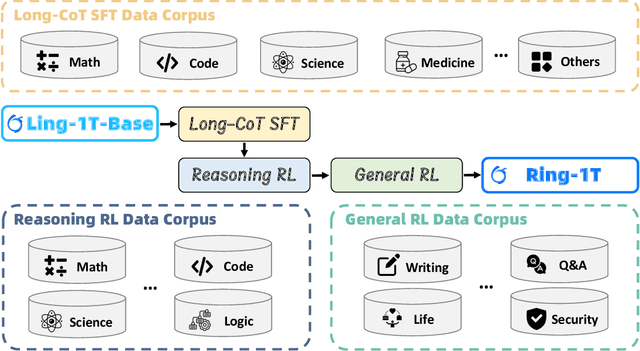
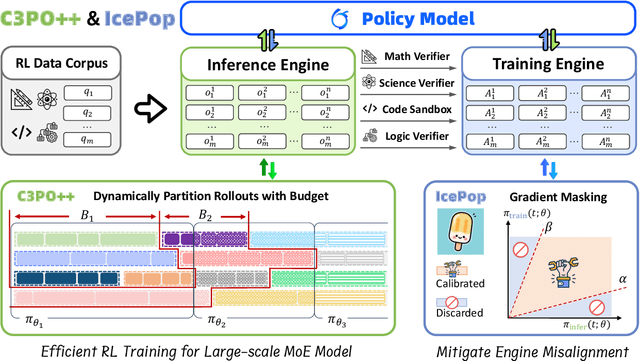
We present Ring-1T, the first open-source, state-of-the-art thinking model with a trillion-scale parameter. It features 1 trillion total parameters and activates approximately 50 billion per token. Training such models at a trillion-parameter scale introduces unprecedented challenges, including train-inference misalignment, inefficiencies in rollout processing, and bottlenecks in the RL system. To address these, we pioneer three interconnected innovations: (1) IcePop stabilizes RL training via token-level discrepancy masking and clipping, resolving instability from training-inference mismatches; (2) C3PO++ improves resource utilization for long rollouts under a token budget by dynamically partitioning them, thereby obtaining high time efficiency; and (3) ASystem, a high-performance RL framework designed to overcome the systemic bottlenecks that impede trillion-parameter model training. Ring-1T delivers breakthrough results across critical benchmarks: 93.4 on AIME-2025, 86.72 on HMMT-2025, 2088 on CodeForces, and 55.94 on ARC-AGI-v1. Notably, it attains a silver medal-level result on the IMO-2025, underscoring its exceptional reasoning capabilities. By releasing the complete 1T parameter MoE model to the community, we provide the research community with direct access to cutting-edge reasoning capabilities. This contribution marks a significant milestone in democratizing large-scale reasoning intelligence and establishes a new baseline for open-source model performance.
MolSnap: Snap-Fast Molecular Generation with Latent Variational Mean Flow
Aug 07, 2025Molecular generation conditioned on textual descriptions is a fundamental task in computational chemistry and drug discovery. Existing methods often struggle to simultaneously ensure high-quality, diverse generation and fast inference. In this work, we propose a novel causality-aware framework that addresses these challenges through two key innovations. First, we introduce a Causality-Aware Transformer (CAT) that jointly encodes molecular graph tokens and text instructions while enforcing causal dependencies during generation. Second, we develop a Variational Mean Flow (VMF) framework that generalizes existing flow-based methods by modeling the latent space as a mixture of Gaussians, enhancing expressiveness beyond unimodal priors. VMF enables efficient one-step inference while maintaining strong generation quality and diversity. Extensive experiments on four standard molecular benchmarks demonstrate that our model outperforms state-of-the-art baselines, achieving higher novelty (up to 74.5\%), diversity (up to 70.3\%), and 100\% validity across all datasets. Moreover, VMF requires only one number of function evaluation (NFE) during conditional generation and up to five NFEs for unconditional generation, offering substantial computational efficiency over diffusion-based methods.
DepMicroDiff: Diffusion-Based Dependency-Aware Multimodal Imputation for Microbiome Data
Jul 31, 2025Microbiome data analysis is essential for understanding host health and disease, yet its inherent sparsity and noise pose major challenges for accurate imputation, hindering downstream tasks such as biomarker discovery. Existing imputation methods, including recent diffusion-based models, often fail to capture the complex interdependencies between microbial taxa and overlook contextual metadata that can inform imputation. We introduce DepMicroDiff, a novel framework that combines diffusion-based generative modeling with a Dependency-Aware Transformer (DAT) to explicitly capture both mutual pairwise dependencies and autoregressive relationships. DepMicroDiff is further enhanced by VAE-based pretraining across diverse cancer datasets and conditioning on patient metadata encoded via a large language model (LLM). Experiments on TCGA microbiome datasets show that DepMicroDiff substantially outperforms state-of-the-art baselines, achieving higher Pearson correlation (up to 0.712), cosine similarity (up to 0.812), and lower RMSE and MAE across multiple cancer types, demonstrating its robustness and generalizability for microbiome imputation.
RefiDiff: Refinement-Aware Diffusion for Efficient Missing Data Imputation
May 20, 2025Missing values in high-dimensional, mixed-type datasets pose significant challenges for data imputation, particularly under Missing Not At Random (MNAR) mechanisms. Existing methods struggle to integrate local and global data characteristics, limiting performance in MNAR and high-dimensional settings. We propose an innovative framework, RefiDiff, combining local machine learning predictions with a novel Mamba-based denoising network capturing interrelationships among distant features and samples. Our approach leverages pre-refinement for initial warm-up imputations and post-refinement to polish results, enhancing stability and accuracy. By encoding mixed-type data into unified tokens, RefiDiff enables robust imputation without architectural or hyperparameter tuning. RefiDiff outperforms state-of-the-art (SOTA) methods across missing-value settings, excelling in MNAR with a 4x faster training time than SOTA DDPM-based approaches. Extensive evaluations on nine real-world datasets demonstrate its robustness, scalability, and effectiveness in handling complex missingness patterns.
TabKAN: Advancing Tabular Data Analysis using Kolmograv-Arnold Network
Apr 09, 2025Tabular data analysis presents unique challenges due to its heterogeneous feature types, missing values, and complex interactions. While traditional machine learning methods, such as gradient boosting, often outperform deep learning approaches, recent advancements in neural architectures offer promising alternatives. This paper introduces TabKAN, a novel framework that advances tabular data modeling using Kolmogorov-Arnold Networks (KANs). Unlike conventional deep learning models, KANs leverage learnable activation functions on edges, enhancing both interpretability and training efficiency. Our contributions include: (1) the introduction of modular KAN-based architectures tailored for tabular data analysis, (2) the development of a transfer learning framework for KAN models, enabling effective knowledge transfer between domains, (3) the development of model-specific interpretability for tabular data learning, reducing reliance on post hoc and model-agnostic analysis, and (4) comprehensive evaluation of vanilla supervised learning across binary and multi-class classification tasks. Through extensive benchmarking on diverse public datasets, TabKAN demonstrates superior performance in supervised learning while significantly outperforming classical and Transformer-based models in transfer learning scenarios. Our findings highlight the advantage of KAN-based architectures in efficiently transferring knowledge across domains, bridging the gap between traditional machine learning and deep learning for structured data.
TabNSA: Native Sparse Attention for Efficient Tabular Data Learning
Mar 12, 2025Tabular data poses unique challenges for deep learning due to its heterogeneous features and lack of inherent spatial structure. This paper introduces TabNSA, a novel deep learning architecture leveraging Native Sparse Attention (NSA) specifically for efficient tabular data processing. TabNSA incorporates a dynamic hierarchical sparse strategy, combining coarse-grained feature compression with fine-grained feature selection to preserve both global context awareness and local precision. By dynamically focusing on relevant subsets of features, TabNSA effectively captures intricate feature interactions. Extensive experiments demonstrate that TabNSA consistently outperforms existing methods, including both deep learning architectures and ensemble decision trees, achieving state-of-the-art performance across various benchmark datasets.
Mol-CADiff: Causality-Aware Autoregressive Diffusion for Molecule Generation
Mar 07, 2025The design of novel molecules with desired properties is a key challenge in drug discovery and materials science. Traditional methods rely on trial-and-error, while recent deep learning approaches have accelerated molecular generation. However, existing models struggle with generating molecules based on specific textual descriptions. We introduce Mol-CADiff, a novel diffusion-based framework that uses causal attention mechanisms for text-conditional molecular generation. Our approach explicitly models the causal relationship between textual prompts and molecular structures, overcoming key limitations in existing methods. We enhance dependency modeling both within and across modalities, enabling precise control over the generation process. Our extensive experiments demonstrate that Mol-CADiff outperforms state-of-the-art methods in generating diverse, novel, and chemically valid molecules, with better alignment to specified properties, enabling more intuitive language-driven molecular design.
CausalGeD: Blending Causality and Diffusion for Spatial Gene Expression Generation
Feb 11, 2025



The integration of single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics (ST) data is crucial for understanding gene expression in spatial context. Existing methods for such integration have limited performance, with structural similarity often below 60\%, We attribute this limitation to the failure to consider causal relationships between genes. We present CausalGeD, which combines diffusion and autoregressive processes to leverage these relationships. By generalizing the Causal Attention Transformer from image generation to gene expression data, our model captures regulatory mechanisms without predefined relationships. Across 10 tissue datasets, CausalGeD outperformed state-of-the-art baselines by 5- 32\% in key metrics, including Pearson's correlation and structural similarity, advancing both technical and biological insights.
PROTECT: Protein circadian time prediction using unsupervised learning
Jan 13, 2025Circadian rhythms regulate the physiology and behavior of humans and animals. Despite advancements in understanding these rhythms and predicting circadian phases at the transcriptional level, predicting circadian phases from proteomic data remains elusive. This challenge is largely due to the scarcity of time labels in proteomic datasets, which are often characterized by small sample sizes, high dimensionality, and significant noise. Furthermore, existing methods for predicting circadian phases from transcriptomic data typically rely on prior knowledge of known rhythmic genes, making them unsuitable for proteomic datasets. To address this gap, we developed a novel computational method using unsupervised deep learning techniques to predict circadian sample phases from proteomic data without requiring time labels or prior knowledge of proteins or genes. Our model involves a two-stage training process optimized for robust circadian phase prediction: an initial greedy one-layer-at-a-time pre-training which generates informative initial parameters followed by fine-tuning. During fine-tuning, a specialized loss function guides the model to align protein expression levels with circadian patterns, enabling it to accurately capture the underlying rhythmic structure within the data. We tested our method on both time-labeled and unlabeled proteomic data. For labeled data, we compared our predictions to the known time labels, achieving high accuracy, while for unlabeled human datasets, including postmortem brain regions and urine samples, we explored circadian disruptions. Notably, our analysis identified disruptions in rhythmic proteins between Alzheimer's disease and control subjects across these samples.
Deep Learning in Single-Cell and Spatial Transcriptomics Data Analysis: Advances and Challenges from a Data Science Perspective
Dec 04, 2024



The development of single-cell and spatial transcriptomics has revolutionized our capacity to investigate cellular properties, functions, and interactions in both cellular and spatial contexts. However, the analysis of single-cell and spatial omics data remains challenging. First, single-cell sequencing data are high-dimensional and sparse, often contaminated by noise and uncertainty, obscuring the underlying biological signals. Second, these data often encompass multiple modalities, including gene expression, epigenetic modifications, and spatial locations. Integrating these diverse data modalities is crucial for enhancing prediction accuracy and biological interpretability. Third, while the scale of single-cell sequencing has expanded to millions of cells, high-quality annotated datasets are still limited. Fourth, the complex correlations of biological tissues make it difficult to accurately reconstruct cellular states and spatial contexts. Traditional feature engineering-based analysis methods struggle to deal with the various challenges presented by intricate biological networks. Deep learning has emerged as a powerful tool capable of handling high-dimensional complex data and automatically identifying meaningful patterns, offering significant promise in addressing these challenges. This review systematically analyzes these challenges and discusses related deep learning approaches. Moreover, we have curated 21 datasets from 9 benchmarks, encompassing 58 computational methods, and evaluated their performance on the respective modeling tasks. Finally, we highlight three areas for future development from a technical, dataset, and application perspective. This work will serve as a valuable resource for understanding how deep learning can be effectively utilized in single-cell and spatial transcriptomics analyses, while inspiring novel approaches to address emerging challenges.