 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDAG Learning from Zero-Inflated Count Data Using Continuous Optimization
Dec 18, 2025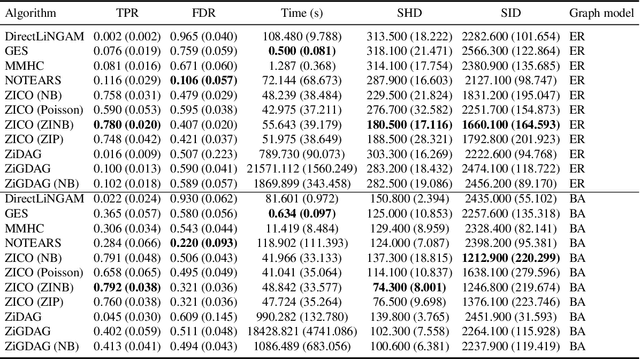
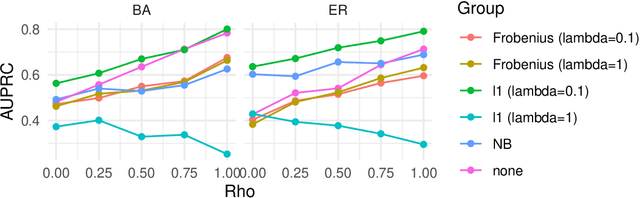
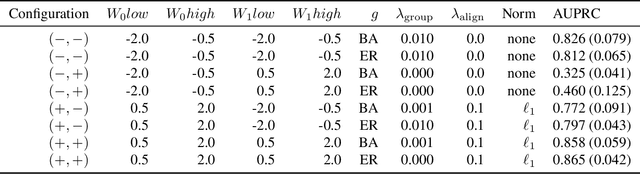
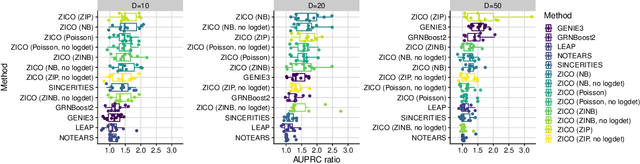
We address network structure learning from zero-inflated count data by casting each node as a zero-inflated generalized linear model and optimizing a smooth, score-based objective under a directed acyclic graph constraint. Our Zero-Inflated Continuous Optimization (ZICO) approach uses node-wise likelihoods with canonical links and enforces acyclicity through a differentiable surrogate constraint combined with sparsity regularization. ZICO achieves superior performance with faster runtimes on simulated data. It also performs comparably to or better than common algorithms for reverse engineering gene regulatory networks. ZICO is fully vectorized and mini-batched, enabling learning on larger variable sets with practical runtimes in a wide range of domains.
Practical Causal Evaluation Metrics for Biological Networks
Nov 16, 2025Estimating causal networks from biological data is a critical step in systems biology. When evaluating the inferred network, assessing the networks based on their intervention effects is particularly important for downstream probabilistic reasoning and the identification of potential drug targets. In the context of gene regulatory network inference, biological databases are often used as reference sources. These databases typically describe relationships in a qualitative rather than quantitative manner. However, few evaluation metrics have been developed that take this qualitative nature into account. To address this, we developed a metric, the sign-augmented Structural Intervention Distance (sSID), and a weighted sSID that incorporates the net effects of the intervention. Through simulations and analyses of real transcriptomic datasets, we found that our proposed metrics could identify a different algorithm as optimal compared to conventional metrics, and the network selected by sSID had a superior performance in the classification task of clinical covariates using transcriptomic data. This suggests that sSID can distinguish networks that are structurally correct but functionally incorrect, highlighting its potential as a more biologically meaningful and practical evaluation metric.
Histo-Genomic Knowledge Distillation For Cancer Prognosis From Histopathology Whole Slide Images
Mar 18, 2024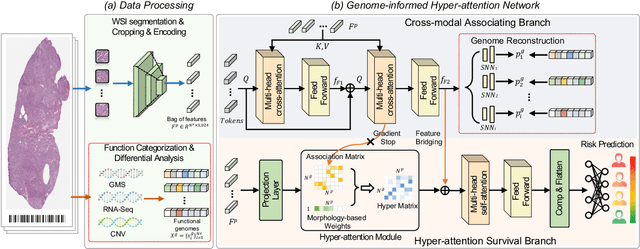
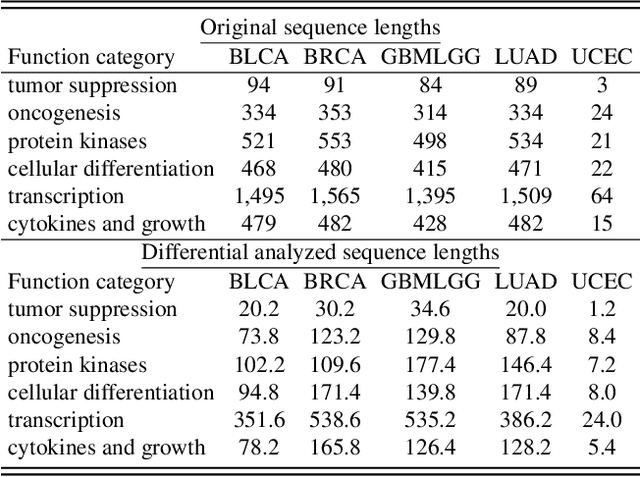
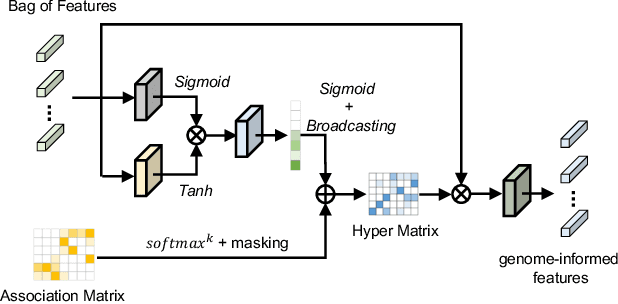
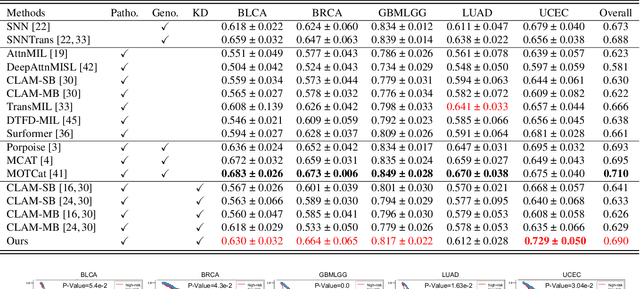
Histo-genomic multi-modal methods have recently emerged as a powerful paradigm, demonstrating significant potential for improving cancer prognosis. However, genome sequencing, unlike histopathology imaging, is still not widely accessible in underdeveloped regions, limiting the application of these multi-modal approaches in clinical settings. To address this, we propose a novel Genome-informed Hyper-Attention Network, termed G-HANet, which is capable of effectively distilling the histo-genomic knowledge during training to elevate uni-modal whole slide image (WSI)-based inference for the first time. Compared with traditional knowledge distillation methods (i.e., teacher-student architecture) in other tasks, our end-to-end model is superior in terms of training efficiency and learning cross-modal interactions. Specifically, the network comprises the cross-modal associating branch (CAB) and hyper-attention survival branch (HSB). Through the genomic data reconstruction from WSIs, CAB effectively distills the associations between functional genotypes and morphological phenotypes and offers insights into the gene expression profiles in the feature space. Subsequently, HSB leverages the distilled histo-genomic associations as well as the generated morphology-based weights to achieve the hyper-attention modeling of the patients from both histopathology and genomic perspectives to improve cancer prognosis. Extensive experiments are conducted on five TCGA benchmarking datasets and the results demonstrate that G-HANet significantly outperforms the state-of-the-art WSI-based methods and achieves competitive performance with genome-based and multi-modal methods. G-HANet is expected to be explored as a useful tool by the research community to address the current bottleneck of insufficient histo-genomic data pairing in the context of cancer prognosis and precision oncology.
Multiplex-detection Based Multiple Instance Learning Network for Whole Slide Image Classification
Aug 06, 2022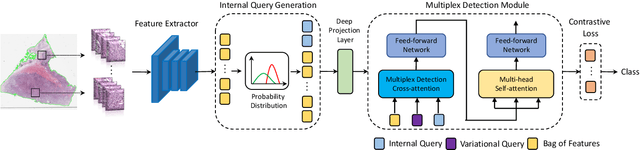
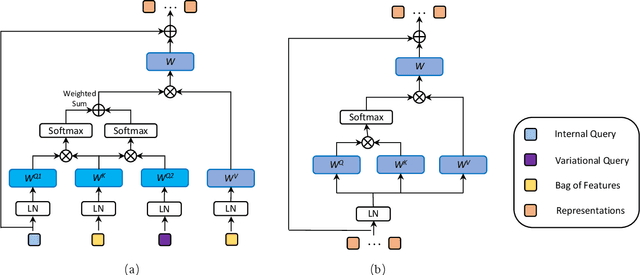
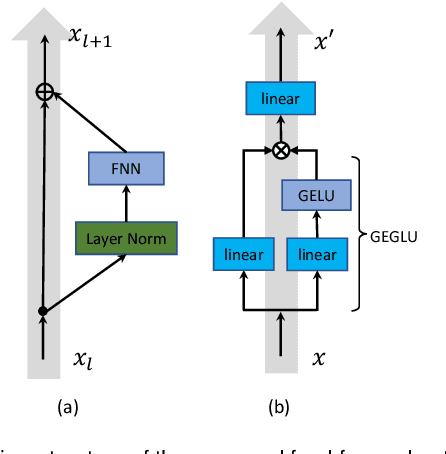
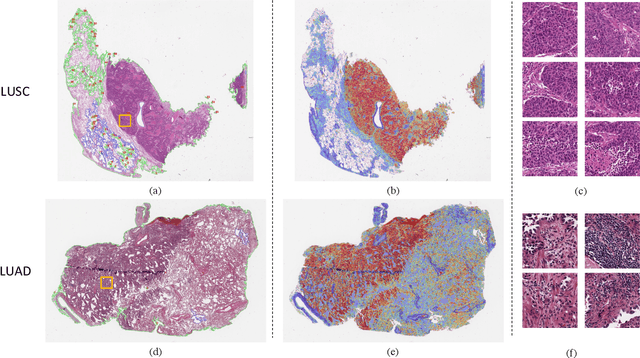
Multiple instance learning (MIL) is a powerful approach to classify whole slide images (WSIs) for diagnostic pathology. A fundamental challenge of MIL on WSI classification is to discover the \textit{critical instances} that trigger the bag label. However, previous methods are primarily designed under the independent and identical distribution hypothesis (\textit{i.i.d}), ignoring either the correlations between instances or heterogeneity of tumours. In this paper, we propose a novel multiplex-detection-based multiple instance learning (MDMIL) to tackle the issues above. Specifically, MDMIL is constructed by the internal query generation module (IQGM) and the multiplex detection module (MDM) and assisted by the memory-based contrastive loss during training. Firstly, IQGM gives the probability of instances and generates the internal query (IQ) for the subsequent MDM by aggregating highly reliable features after the distribution analysis. Secondly, the multiplex-detection cross-attention (MDCA) and multi-head self-attention (MHSA) in MDM cooperate to generate the final representations for the WSI. In this process, the IQ and trainable variational query (VQ) successfully build up the connections between instances and significantly improve the model's robustness toward heterogeneous tumours. At last, to further enforce constraints in the feature space and stabilize the training process, we adopt a memory-based contrastive loss, which is practicable for WSI classification even with a single sample as input in each iteration. We conduct experiments on three computational pathology datasets, e.g., CAMELYON16, TCGA-NSCLC, and TCGA-RCC datasets. The superior accuracy and AUC demonstrate the superiority of our proposed MDMIL over other state-of-the-art methods.
Finding Exogenous Variables in Data with Many More Variables than Observations
Apr 07, 2011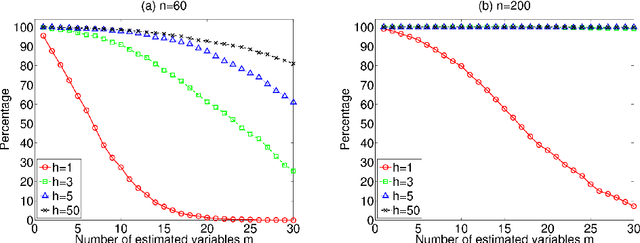
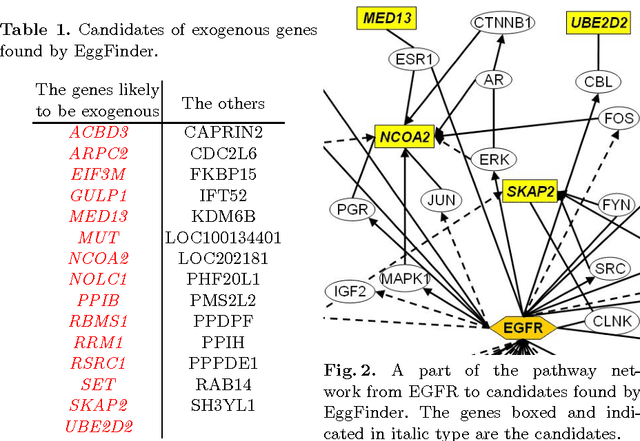
Many statistical methods have been proposed to estimate causal models in classical situations with fewer variables than observations (p<n, p: the number of variables and n: the number of observations). However, modern datasets including gene expression data need high-dimensional causal modeling in challenging situations with orders of magnitude more variables than observations (p>>n). In this paper, we propose a method to find exogenous variables in a linear non-Gaussian causal model, which requires much smaller sample sizes than conventional methods and works even when p>>n. The key idea is to identify which variables are exogenous based on non-Gaussianity instead of estimating the entire structure of the model. Exogenous variables work as triggers that activate a causal chain in the model, and their identification leads to more efficient experimental designs and better understanding of the causal mechanism. We present experiments with artificial data and real-world gene expression data to evaluate the method.
* A revised version of this was published in Proc. ICANN2010