 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRiemannian Neural Optimal Transport
Feb 03, 2026Computational optimal transport (OT) offers a principled framework for generative modeling. Neural OT methods, which use neural networks to learn an OT map (or potential) from data in an amortized way, can be evaluated out of sample after training, but existing approaches are tailored to Euclidean geometry. Extending neural OT to high-dimensional Riemannian manifolds remains an open challenge. In this paper, we prove that any method for OT on manifolds that produces discrete approximations of transport maps necessarily suffers from the curse of dimensionality: achieving a fixed accuracy requires a number of parameters that grows exponentially with the manifold dimension. Motivated by this limitation, we introduce Riemannian Neural OT (RNOT) maps, which are continuous neural-network parameterizations of OT maps on manifolds that avoid discretization and incorporate geometric structure by construction. Under mild regularity assumptions, we prove that RNOT maps approximate Riemannian OT maps with sub-exponential complexity in the dimension. Experiments on synthetic and real datasets demonstrate improved scalability and competitive performance relative to discretization-based baselines.
Interpretable Graph Learning Over Sets of Temporally-Sparse Data
May 25, 2025Real-world medical data often includes measurements from multiple signals that are collected at irregular and asynchronous time intervals. For example, different types of blood tests can be measured at different times and frequencies, resulting in fragmented and unevenly scattered temporal data. Similar issues of irregular sampling of different attributes occur in other domains, such as monitoring of large systems using event log files or the spread of fake news on social networks. Effectively learning from such data requires models that can handle sets of temporally sparse and heterogeneous signals. In this paper, we propose Graph Mixing Additive Networks (GMAN), a novel and interpretable-by-design model for learning over irregular sets of temporal signals. Our method achieves state-of-the-art performance in real-world medical tasks, including a 4-point increase in the AUROC score of in-hospital mortality prediction, compared to existing methods. We further showcase GMAN's flexibility by applying it to a fake news detection task. We demonstrate how its interpretability capabilities, including node-level, graph-level, and subset-level importance, allow for transition phases detection and gaining medical insights with real-world high-stakes implications. Finally, we provide theoretical insights on GMAN expressive power.
NeuralSurv: Deep Survival Analysis with Bayesian Uncertainty Quantification
May 16, 2025We introduce NeuralSurv, the first deep survival model to incorporate Bayesian uncertainty quantification. Our non-parametric, architecture-agnostic framework flexibly captures time-varying covariate-risk relationships in continuous time via a novel two-stage data-augmentation scheme, for which we establish theoretical guarantees. For efficient posterior inference, we introduce a mean-field variational algorithm with coordinate-ascent updates that scale linearly in model size. By locally linearizing the Bayesian neural network, we obtain full conjugacy and derive all coordinate updates in closed form. In experiments, NeuralSurv delivers superior calibration compared to state-of-the-art deep survival models, while matching or exceeding their discriminative performance across both synthetic benchmarks and real-world datasets. Our results demonstrate the value of Bayesian principles in data-scarce regimes by enhancing model calibration and providing robust, well-calibrated uncertainty estimates for the survival function.
Diffusion Models for Inverse Problems in the Exponential Family
Feb 09, 2025Diffusion models have emerged as powerful tools for solving inverse problems, yet prior work has primarily focused on observations with Gaussian measurement noise, restricting their use in real-world scenarios. This limitation persists due to the intractability of the likelihood score, which until now has only been approximated in the simpler case of Gaussian likelihoods. In this work, we extend diffusion models to handle inverse problems where the observations follow a distribution from the exponential family, such as a Poisson or a Binomial distribution. By leveraging the conjugacy properties of exponential family distributions, we introduce the evidence trick, a method that provides a tractable approximation to the likelihood score. In our experiments, we demonstrate that our methodology effectively performs Bayesian inference on spatially inhomogeneous Poisson processes with intensities as intricate as ImageNet images. Furthermore, we demonstrate the real-world impact of our methodology by showing that it performs competitively with the current state-of-the-art in predicting malaria prevalence estimates in Sub-Saharan Africa.
Contrastive Deep Learning Reveals Age Biomarkers in Histopathological Skin Biopsies
Nov 25, 2024
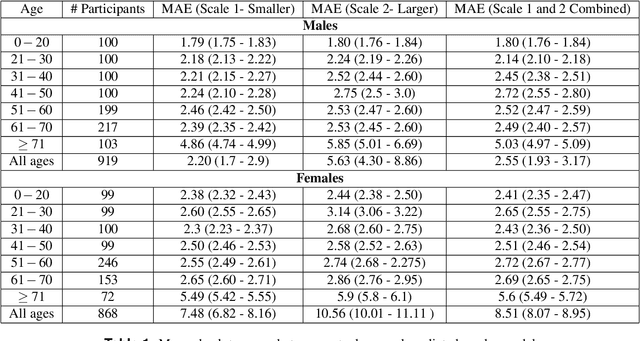


As global life expectancy increases, so does the burden of chronic diseases, yet individuals exhibit considerable variability in the rate at which they age. Identifying biomarkers that distinguish fast from slow ageing is crucial for understanding the biology of ageing, enabling early disease detection, and improving prevention strategies. Using contrastive deep learning, we show that skin biopsy images alone are sufficient to determine an individual's age. We then use visual features in histopathology slides of the skin biopsies to construct a novel biomarker of ageing. By linking with comprehensive health registers in Denmark, we demonstrate that visual features in histopathology slides of skin biopsies predict mortality and the prevalence of chronic age-related diseases. Our work highlights how routinely collected health data can provide additional value when used together with deep learning, by creating a new biomarker for ageing which can be actively used to determine mortality over time.
KidSat: satellite imagery to map childhood poverty dataset and benchmark
Jul 08, 2024


Satellite imagery has emerged as an important tool to analyse demographic, health, and development indicators. While various deep learning models have been built for these tasks, each is specific to a particular problem, with few standard benchmarks available. We propose a new dataset pairing satellite imagery and high-quality survey data on child poverty to benchmark satellite feature representations. Our dataset consists of 33,608 images, each 10 km $\times$ 10 km, from 19 countries in Eastern and Southern Africa in the time period 1997-2022. As defined by UNICEF, multidimensional child poverty covers six dimensions and it can be calculated from the face-to-face Demographic and Health Surveys (DHS) Program . As part of the benchmark, we test spatial as well as temporal generalization, by testing on unseen locations, and on data after the training years. Using our dataset we benchmark multiple models, from low-level satellite imagery models such as MOSAIKS , to deep learning foundation models, which include both generic vision models such as Self-Distillation with no Labels (DINOv2) models and specific satellite imagery models such as SatMAE. We provide open source code for building the satellite dataset, obtaining ground truth data from DHS and running various models assessed in our work.
Continuous football player tracking from discrete broadcast data
Nov 24, 2023


Player tracking data remains out of reach for many professional football teams as their video feeds are not sufficiently high quality for computer vision technologies to be used. To help bridge this gap, we present a method that can estimate continuous full-pitch tracking data from discrete data made from broadcast footage. Such data could be collected by clubs or players at a similar cost to event data, which is widely available down to semi-professional level. We test our method using open-source tracking data, and include a version that can be applied to a large set of over 200 games with such discrete data.
Leaping through tree space: continuous phylogenetic inference for rooted and unrooted trees
Jun 15, 2023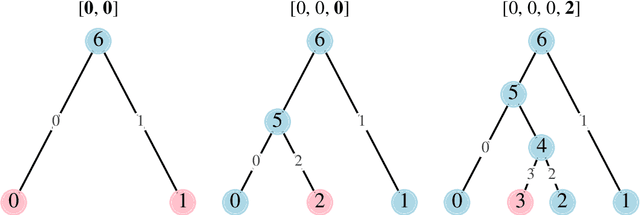
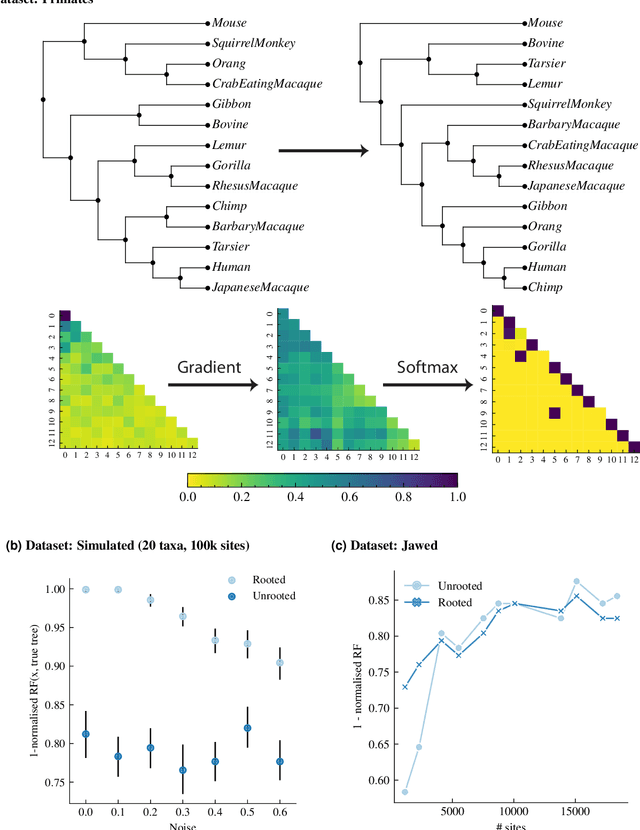
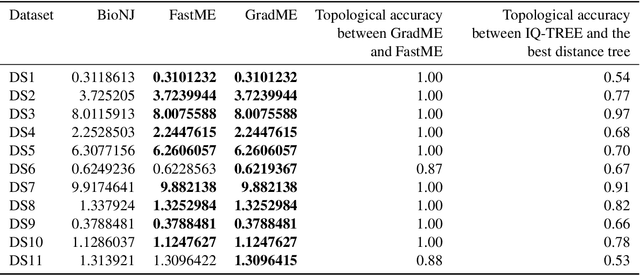
Phylogenetics is now fundamental in life sciences, providing insights into the earliest branches of life and the origins and spread of epidemics. However, finding suitable phylogenies from the vast space of possible trees remains challenging. To address this problem, for the first time, we perform both tree exploration and inference in a continuous space where the computation of gradients is possible. This continuous relaxation allows for major leaps across tree space in both rooted and unrooted trees, and is less susceptible to convergence to local minima. Our approach outperforms the current best methods for inference on unrooted trees and, in simulation, accurately infers the tree and root in ultrametric cases. The approach is effective in cases of empirical data with negligible amounts of data, which we demonstrate on the phylogeny of jawed vertebrates. Indeed, only a few genes with an ultrametric signal were generally sufficient for resolving the major lineages of vertebrate. With cubic-time complexity and efficient optimisation via automatic differentiation, our method presents an effective way forwards for exploring the most difficult, data-deficient phylogenetic questions.
Deep learning and MCMC with aggVAE for shifting administrative boundaries: mapping malaria prevalence in Kenya
May 31, 2023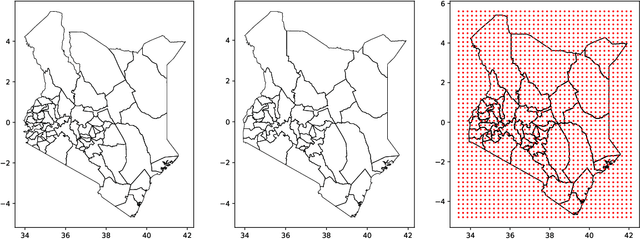
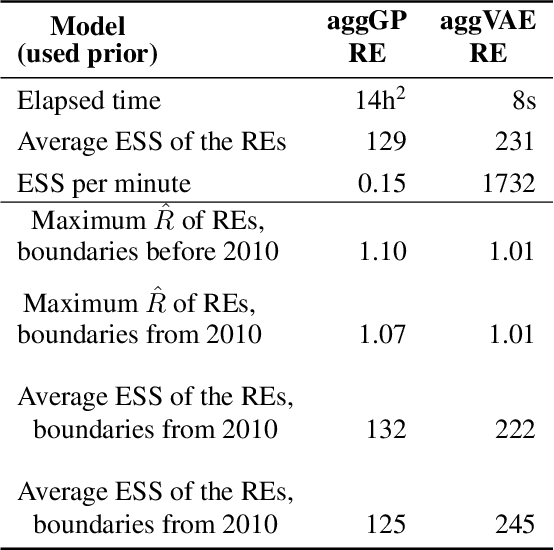
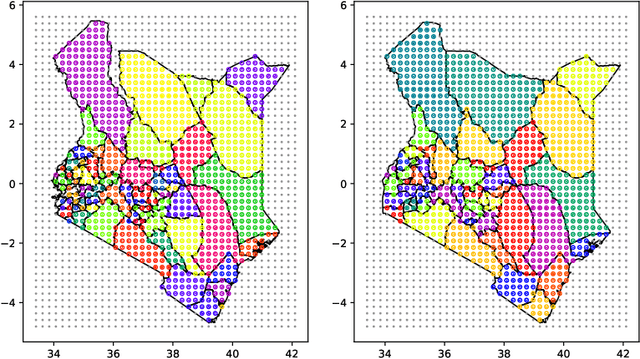

Model-based disease mapping remains a fundamental policy-informing tool in public health and disease surveillance with hierarchical Bayesian models being the current state-of-the-art approach. When working with areal data, e.g. aggregates at the administrative unit level such as district or province, routinely used models rely on the adjacency structure of areal units to account for spatial correlations. The goal of disease surveillance systems is to track disease outcomes over time, but this provides challenging in situations of crises, such as political changes, leading to changes of administrative boundaries. Kenya is an example of such country. Moreover, adjacency-based approach ignores the continuous nature of spatial processes and cannot solve the change-of-support problem, i.e. when administrative boundaries change. We present a novel, practical, and easy to implement solution relying on a methodology combining deep generative modelling and fully Bayesian inference. We build on the recent work of PriorVAE able to encode spatial priors over small areas with variational autoencoders, to map malaria prevalence in Kenya. We solve the change-of-support problem arising from Kenya changing its district boundaries in 2010. We draw realisations of the Gaussian Process (GP) prior over a fine artificial spatial grid representing continuous space and then aggregate these realisations to the level of administrative boundaries. The aggregated values are then encoded using the PriorVAE technique. The trained priors (aggVAE) are then used at the inference stage instead of the GP priors within a Markov chain Monte Carlo (MCMC) scheme. We demonstrate that it is possible to use the flexible and appropriate model for areal data based on aggregation of continuous priors, and that inference is orders of magnitude faster when using aggVAE than combining the original GP priors and the aggregation step.
A comparison of short-term probabilistic forecasts for the incidence of COVID-19 using mechanistic and statistical time series models
May 01, 2023Short-term forecasts of infectious disease spread are a critical component in risk evaluation and public health decision making. While different models for short-term forecasting have been developed, open questions about their relative performance remain. Here, we compare short-term probabilistic forecasts of popular mechanistic models based on the renewal equation with forecasts of statistical time series models. Our empirical comparison is based on data of the daily incidence of COVID-19 across six large US states over the first pandemic year. We find that, on average, probabilistic forecasts from statistical time series models are overall at least as accurate as forecasts from mechanistic models. Moreover, statistical time series models better capture volatility. Our findings suggest that domain knowledge, which is integrated into mechanistic models by making assumptions about disease dynamics, does not improve short-term forecasts of disease incidence. We note, however, that forecasting is often only one of many objectives and thus mechanistic models remain important, for example, to model the impact of vaccines or the emergence of new variants.