 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting Genetic Mutations from Single-Cell Bone Marrow Images in Acute Myeloid Leukemia Using Noise-Robust Deep Learning Models
Jun 15, 2025In this study, we propose a robust methodology for identification of myeloid blasts followed by prediction of genetic mutation in single-cell images of blasts, tackling challenges associated with label accuracy and data noise. We trained an initial binary classifier to distinguish between leukemic (blasts) and non-leukemic cells images, achieving 90 percent accuracy. To evaluate the models generalization, we applied this model to a separate large unlabeled dataset and validated the predictions with two haemato-pathologists, finding an approximate error rate of 20 percent in the leukemic and non-leukemic labels. Assuming this level of label noise, we further trained a four-class model on images predicted as blasts to classify specific mutations. The mutation labels were known for only a bag of cell images extracted from a single slide. Despite the tumor label noise, our mutation classification model achieved 85 percent accuracy across four mutation classes, demonstrating resilience to label inconsistencies. This study highlights the capability of machine learning models to work with noisy labels effectively while providing accurate, clinically relevant mutation predictions, which is promising for diagnostic applications in areas such as haemato-pathology.
Scalable Whole Slide Image Representation Using K-Mean Clustering and Fisher Vector Aggregation
Jan 21, 2025


Whole slide images (WSIs) are high-resolution, gigapixel sized images that pose significant computational challenges for traditional machine learning models due to their size and heterogeneity.In this paper, we present a scalable and efficient methodology for WSI classification by leveraging patch-based feature extraction, clustering, and Fisher vector encoding. Initially, WSIs are divided into fixed size patches, and deep feature embeddings are extracted from each patch using a pre-trained convolutional neural network (CNN). These patch-level embeddings are subsequently clustered using K-means clustering, where each cluster aggregates semantically similar regions of the WSI. To effectively summarize each cluster, Fisher vector representations are computed by modeling the distribution of patch embeddings in each cluster as a parametric Gaussian mixture model (GMM). The Fisher vectors from each cluster are concatenated into a high-dimensional feature vector, creating a compact and informative representation of the entire WSI. This feature vector is then used by a classifier to predict the WSI's diagnostic label. Our method captures local and global tissue structures and yields robust performance for large-scale WSI classification, demonstrating superior accuracy and scalability compared to other approaches.
Clustered Patch Embeddings for Permutation-Invariant Classification of Whole Slide Images
Nov 13, 2024


Whole Slide Imaging (WSI) is a cornerstone of digital pathology, offering detailed insights critical for diagnosis and research. Yet, the gigapixel size of WSIs imposes significant computational challenges, limiting their practical utility. Our novel approach addresses these challenges by leveraging various encoders for intelligent data reduction and employing a different classification model to ensure robust, permutation-invariant representations of WSIs. A key innovation of our method is the ability to distill the complex information of an entire WSI into a single vector, effectively capturing the essential features needed for accurate analysis. This approach significantly enhances the computational efficiency of WSI analysis, enabling more accurate pathological assessments without the need for extensive computational resources. This breakthrough equips us with the capability to effectively address the challenges posed by large image resolutions in whole-slide imaging, paving the way for more scalable and effective utilization of WSIs in medical diagnostics and research, marking a significant advancement in the field.
Efficient Whole Slide Image Classification through Fisher Vector Representation
Nov 13, 2024



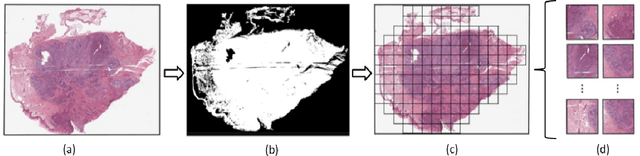
The advancement of digital pathology, particularly through computational analysis of whole slide images (WSI), is poised to significantly enhance diagnostic precision and efficiency. However, the large size and complexity of WSIs make it difficult to analyze and classify them using computers. This study introduces a novel method for WSI classification by automating the identification and examination of the most informative patches, thus eliminating the need to process the entire slide. Our method involves two-stages: firstly, it extracts only a few patches from the WSIs based on their pathological significance; and secondly, it employs Fisher vectors (FVs) for representing features extracted from these patches, which is known for its robustness in capturing fine-grained details. This approach not only accentuates key pathological features within the WSI representation but also significantly reduces computational overhead, thus making the process more efficient and scalable. We have rigorously evaluated the proposed method across multiple datasets to benchmark its performance against comprehensive WSI analysis and contemporary weakly-supervised learning methodologies. The empirical results indicate that our focused analysis of select patches, combined with Fisher vector representation, not only aligns with, but at times surpasses, the classification accuracy of standard practices. Moreover, this strategy notably diminishes computational load and resource expenditure, thereby establishing an efficient and precise framework for WSI analysis in the realm of digital pathology.
Classification and Morphological Analysis of DLBCL Subtypes in H\&E-Stained Slides
Nov 13, 2024
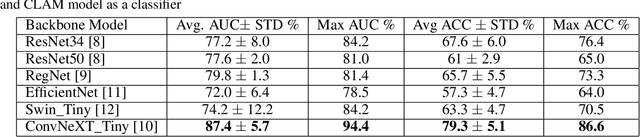
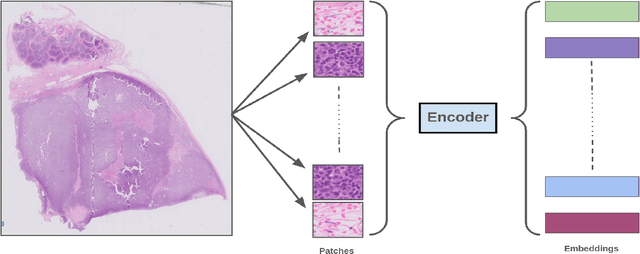
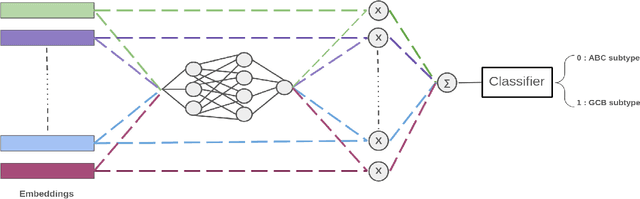
We address the challenge of automated classification of diffuse large B-cell lymphoma (DLBCL) into its two primary subtypes: activated B-cell-like (ABC) and germinal center B-cell-like (GCB). Accurate classification between these subtypes is essential for determining the appropriate therapeutic strategy, given their distinct molecular profiles and treatment responses. Our proposed deep learning model demonstrates robust performance, achieving an average area under the curve (AUC) of (87.4 pm 5.7)\% during cross-validation. It shows a high positive predictive value (PPV), highlighting its potential for clinical application, such as triaging for molecular testing. To gain biological insights, we performed an analysis of morphological features of ABC and GCB subtypes. We segmented cell nuclei using a pre-trained deep neural network and compared the statistics of geometric and color features for ABC and GCB. We found that the distributions of these features were not very different for the two subtypes, which suggests that the visual differences between them are more subtle. These results underscore the potential of our method to assist in more precise subtype classification and can contribute to improved treatment management and outcomes for patients of DLBCL.
Cross-Domain Evaluation of Few-Shot Classification Models: Natural Images vs. Histopathological Images
Oct 11, 2024


In this study, we investigate the performance of few-shot classification models across different domains, specifically natural images and histopathological images. We first train several few-shot classification models on natural images and evaluate their performance on histopathological images. Subsequently, we train the same models on histopathological images and compare their performance. We incorporated four histopathology datasets and one natural images dataset and assessed performance across 5-way 1-shot, 5-way 5-shot, and 5-way 10-shot scenarios using a selection of state-of-the-art classification techniques. Our experimental results reveal insights into the transferability and generalization capabilities of few-shot classification models between diverse image domains. We analyze the strengths and limitations of these models in adapting to new domains and provide recommendations for optimizing their performance in cross-domain scenarios. This research contributes to advancing our understanding of few-shot learning in the context of image classification across diverse domains.
HER2 and FISH Status Prediction in Breast Biopsy H&E-Stained Images Using Deep Learning
Aug 25, 2024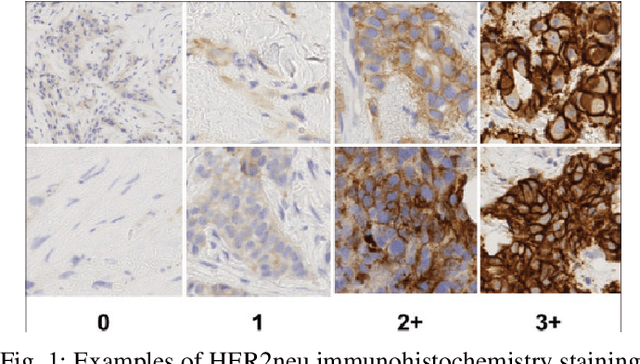
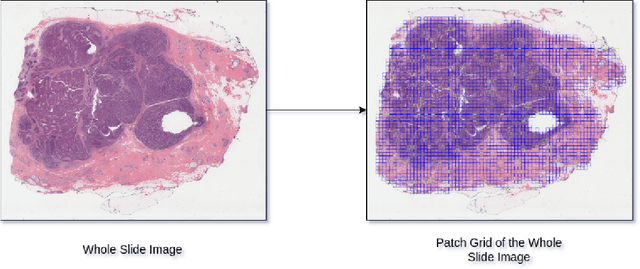

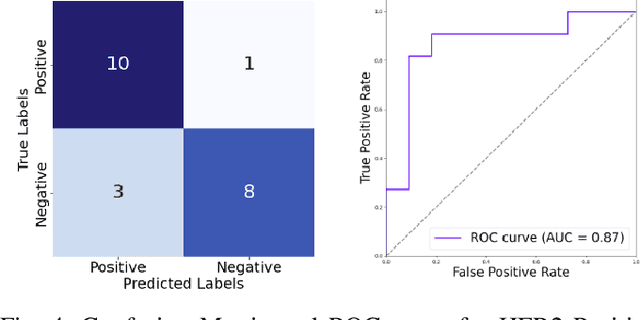
The current standard for detecting human epidermal growth factor receptor 2 (HER2) status in breast cancer patients relies on HER2 amplification, identified through fluorescence in situ hybridization (FISH) or immunohistochemistry (IHC). However, hematoxylin and eosin (H\&E) tumor stains are more widely available, and accurately predicting HER2 status using H\&E could reduce costs and expedite treatment selection. Deep Learning algorithms for H&E have shown effectiveness in predicting various cancer features and clinical outcomes, including moderate success in HER2 status prediction. In this work, we employed a customized weak supervision classification technique combined with MoCo-v2 contrastive learning to predict HER2 status. We trained our pipeline on 182 publicly available H&E Whole Slide Images (WSIs) from The Cancer Genome Atlas (TCGA), for which annotations by the pathology team at Yale School of Medicine are publicly available. Our pipeline achieved an Area Under the Curve (AUC) of 0.85 across four different test folds. Additionally, we tested our model on 44 H&E slides from the TCGA-BRCA dataset, which had an HER2 score of 2+ and included corresponding HER2 status and FISH test results. These cases are considered equivocal for IHC, requiring an expensive FISH test on their IHC slides for disambiguation. Our pipeline demonstrated an AUC of 0.81 on these challenging H&E slides. Reducing the need for FISH test can have significant implications in cancer treatment equity for underserved populations.
Few-Shot Histopathology Image Classification: Evaluating State-of-the-Art Methods and Unveiling Performance Insights
Aug 25, 2024



This paper presents a study on few-shot classification in the context of histopathology images. While few-shot learning has been studied for natural image classification, its application to histopathology is relatively unexplored. Given the scarcity of labeled data in medical imaging and the inherent challenges posed by diverse tissue types and data preparation techniques, this research evaluates the performance of state-of-the-art few-shot learning methods for various scenarios on histology data. We have considered four histopathology datasets for few-shot histopathology image classification and have evaluated 5-way 1-shot, 5-way 5-shot and 5-way 10-shot scenarios with a set of state-of-the-art classification techniques. The best methods have surpassed an accuracy of 70%, 80% and 85% in the cases of 5-way 1-shot, 5-way 5-shot and 5-way 10-shot cases, respectively. We found that for histology images popular meta-learning approaches is at par with standard fine-tuning and regularization methods. Our experiments underscore the challenges of working with images from different domains and underscore the significance of unbiased and focused evaluations in advancing computer vision techniques for specialized domains, such as histology images.
Advancing Gene Selection in Oncology: A Fusion of Deep Learning and Sparsity for Precision Gene Selection
Mar 04, 2024



Gene selection plays a pivotal role in oncology research for improving outcome prediction accuracy and facilitating cost-effective genomic profiling for cancer patients. This paper introduces two gene selection strategies for deep learning-based survival prediction models. The first strategy uses a sparsity-inducing method while the second one uses importance based gene selection for identifying relevant genes. Our overall approach leverages the power of deep learning to model complex biological data structures, while sparsity-inducing methods ensure the selection process focuses on the most informative genes, minimizing noise and redundancy. Through comprehensive experimentation on diverse genomic and survival datasets, we demonstrate that our strategy not only identifies gene signatures with high predictive power for survival outcomes but can also streamlines the process for low-cost genomic profiling. The implications of this research are profound as it offers a scalable and effective tool for advancing personalized medicine and targeted cancer therapies. By pushing the boundaries of gene selection methodologies, our work contributes significantly to the ongoing efforts in cancer genomics, promising improved diagnostic and prognostic capabilities in clinical settings.
Combining Datasets with Different Label Sets for Improved Nucleus Segmentation and Classification
Oct 05, 2023



Segmentation and classification of cell nuclei in histopathology images using deep neural networks (DNNs) can save pathologists' time for diagnosing various diseases, including cancers, by automating cell counting and morphometric assessments. It is now well-known that the accuracy of DNNs increases with the sizes of annotated datasets available for training. Although multiple datasets of histopathology images with nuclear annotations and class labels have been made publicly available, the set of class labels differ across these datasets. We propose a method to train DNNs for instance segmentation and classification on multiple datasets where the set of classes across the datasets are related but not the same. Specifically, our method is designed to utilize a coarse-to-fine class hierarchy, where the set of classes labeled and annotated in a dataset can be at any level of the hierarchy, as long as the classes are mutually exclusive. Within a dataset, the set of classes need not even be at the same level of the class hierarchy tree. Our results demonstrate that segmentation and classification metrics for the class set used by the test split of a dataset can improve by pre-training on another dataset that may even have a different set of classes due to the expansion of the training set enabled by our method. Furthermore, generalization to previously unseen datasets also improves by combining multiple other datasets with different sets of classes for training. The improvement is both qualitative and quantitative. The proposed method can be adapted for various loss functions, DNN architectures, and application domains.