 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnhanced Prediction of CAR T-Cell Cytotoxicity with Quantum-Kernel Methods
Jul 30, 2025Chimeric antigen receptor (CAR) T-cells are T-cells engineered to recognize and kill specific tumor cells. Through their extracellular domains, CAR T-cells bind tumor cell antigens which triggers CAR T activation and proliferation. These processes are regulated by co-stimulatory domains present in the intracellular region of the CAR T-cell. Through integrating novel signaling components into the co-stimulatory domains, it is possible to modify CAR T-cell phenotype. Identifying and experimentally testing new CAR constructs based on libraries of co-stimulatory domains is nontrivial given the vast combinatorial space defined by such libraries. This leads to a highly data constrained, poorly explored combinatorial problem, where the experiments undersample all possible combinations. We propose a quantum approach using a Projected Quantum Kernel (PQK) to address this challenge. PQK operates by embedding classical data into a high dimensional Hilbert space and employs a kernel method to measure sample similarity. Using 61 qubits on a gate-based quantum computer, we demonstrate the largest PQK application to date and an enhancement in the classification performance over purely classical machine learning methods for CAR T cytotoxicity prediction. Importantly, we show improved learning for specific signaling domains and domain positions, particularly where there was lower information highlighting the potential for quantum computing in data-constrained problems.
Towards Quantum Tensor Decomposition in Biomedical Applications
Feb 19, 2025


Tensor decomposition has emerged as a powerful framework for feature extraction in multi-modal biomedical data. In this review, we present a comprehensive analysis of tensor decomposition methods such as Tucker, CANDECOMP/PARAFAC, spiked tensor decomposition, etc. and their diverse applications across biomedical domains such as imaging, multi-omics, and spatial transcriptomics. To systematically investigate the literature, we applied a topic modeling-based approach that identifies and groups distinct thematic sub-areas in biomedicine where tensor decomposition has been used, thereby revealing key trends and research directions. We evaluated challenges related to the scalability of latent spaces along with obtaining the optimal rank of the tensor, which often hinder the extraction of meaningful features from increasingly large and complex datasets. Additionally, we discuss recent advances in quantum algorithms for tensor decomposition, exploring how quantum computing can be leveraged to address these challenges. Our study includes a preliminary resource estimation analysis for quantum computing platforms and examines the feasibility of implementing quantum-enhanced tensor decomposition methods on near-term quantum devices. Collectively, this review not only synthesizes current applications and challenges of tensor decomposition in biomedical analyses but also outlines promising quantum computing strategies to enhance its impact on deriving actionable insights from complex biomedical data.
Inferring COVID-19 Biological Pathways from Clinical Phenotypes via Topological Analysis
Jan 19, 2021
COVID-19 has caused thousands of deaths around the world and also resulted in a large international economic disruption. Identifying the pathways associated with this illness can help medical researchers to better understand the properties of the condition. This process can be carried out by analyzing the medical records. It is crucial to develop tools and models that can aid researchers with this process in a timely manner. However, medical records are often unstructured clinical notes, and this poses significant challenges to developing the automated systems. In this article, we propose a pipeline to aid practitioners in analyzing clinical notes and revealing the pathways associated with this disease. Our pipeline relies on topological properties and consists of three steps: 1) pre-processing the clinical notes to extract the salient concepts, 2) constructing a feature space of the patients to characterize the extracted concepts, and finally, 3) leveraging the topological properties to distill the available knowledge and visualize the result. Our experiments on a publicly available dataset of COVID-19 clinical notes testify that our pipeline can indeed extract meaningful pathways.
Utilizing stability criteria in choosing feature selection methods yields reproducible results in microbiome data
Nov 30, 2020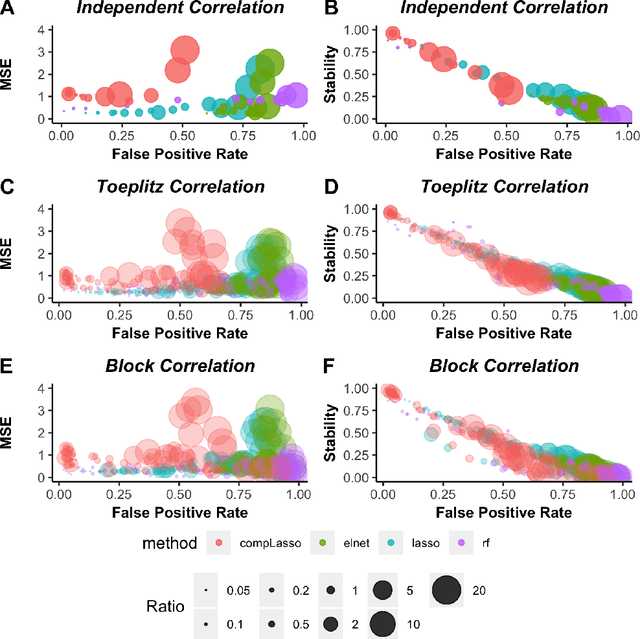
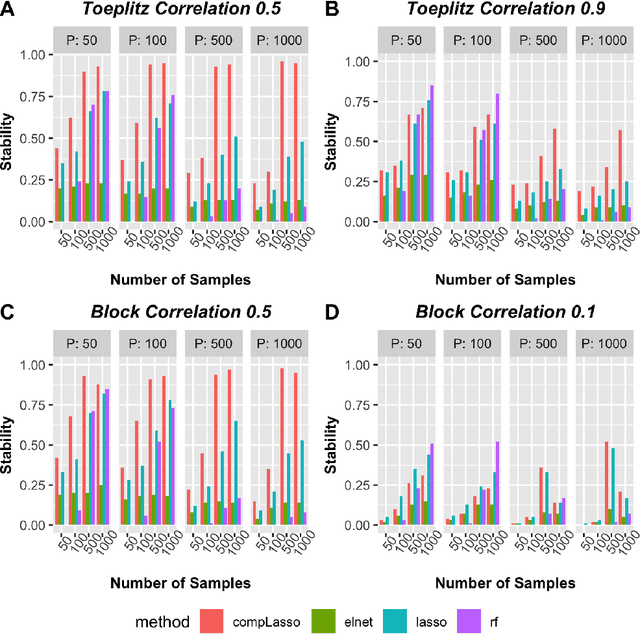

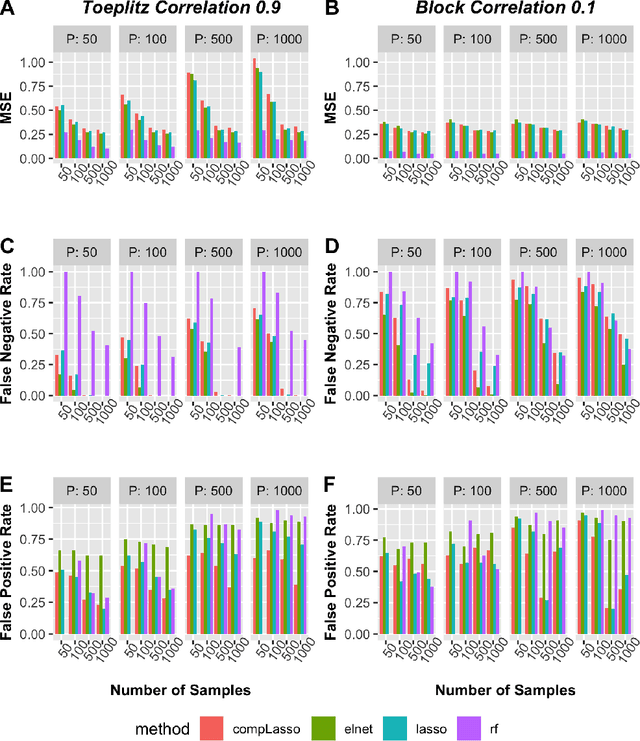
Feature selection is indispensable in microbiome data analysis, but it can be particularly challenging as microbiome data sets are high-dimensional, underdetermined, sparse and compositional. Great efforts have recently been made on developing new methods for feature selection that handle the above data characteristics, but almost all methods were evaluated based on performance of model predictions. However, little attention has been paid to address a fundamental question: how appropriate are those evaluation criteria? Most feature selection methods often control the model fit, but the ability to identify meaningful subsets of features cannot be evaluated simply based on the prediction accuracy. If tiny changes to the training data would lead to large changes in the chosen feature subset, then many of the biological features that an algorithm has found are likely to be a data artifact rather than real biological signal. This crucial need of identifying relevant and reproducible features motivated the reproducibility evaluation criterion such as Stability, which quantifies how robust a method is to perturbations in the data. In our paper, we compare the performance of popular model prediction metric MSE and proposed reproducibility criterion Stability in evaluating four widely used feature selection methods in both simulations and experimental microbiome applications. We conclude that Stability is a preferred feature selection criterion over MSE because it better quantifies the reproducibility of the feature selection method.
MINT: Mutual Information based Transductive Feature Selection for Genetic Trait Prediction
Oct 07, 2013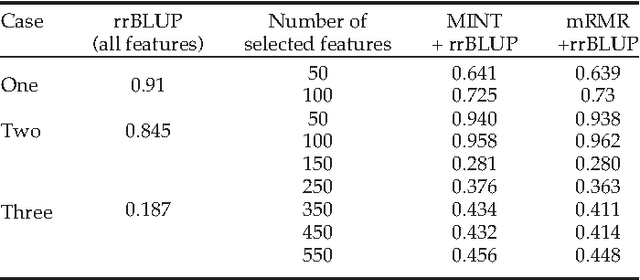
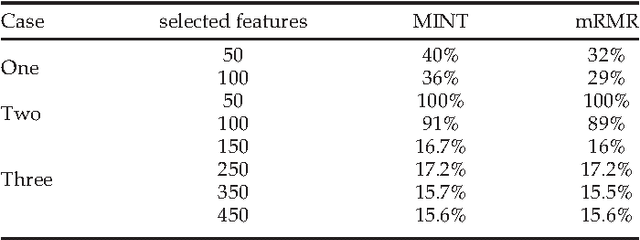
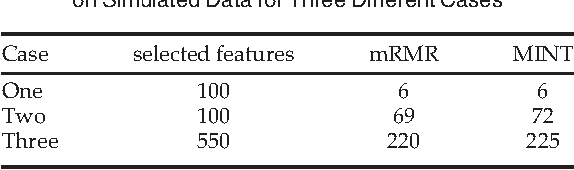

Whole genome prediction of complex phenotypic traits using high-density genotyping arrays has attracted a great deal of attention, as it is relevant to the fields of plant and animal breeding and genetic epidemiology. As the number of genotypes is generally much bigger than the number of samples, predictive models suffer from the curse-of-dimensionality. The curse-of-dimensionality problem not only affects the computational efficiency of a particular genomic selection method, but can also lead to poor performance, mainly due to correlation among markers. In this work we proposed the first transductive feature selection method based on the MRMR (Max-Relevance and Min-Redundancy) criterion which we call MINT. We applied MINT on genetic trait prediction problems and showed that in general MINT is a better feature selection method than the state-of-the-art inductive method mRMR.