 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeaLLoyM: A large language model for alloy phase diagram prediction
Jul 30, 2025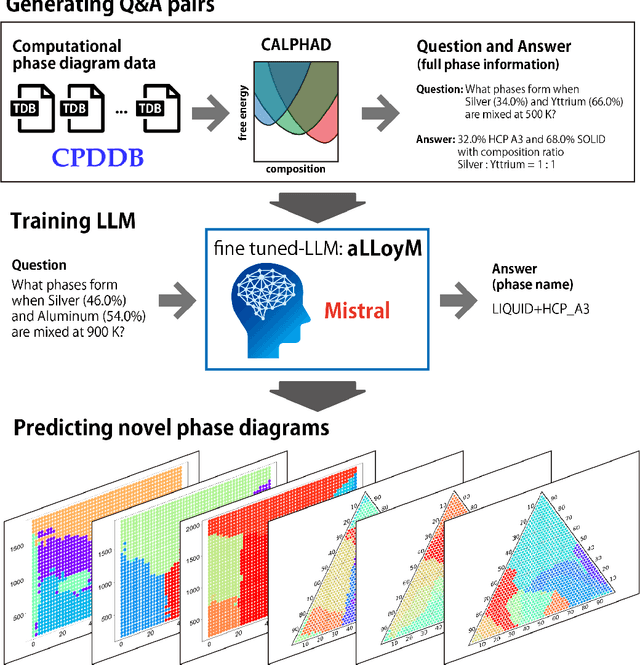
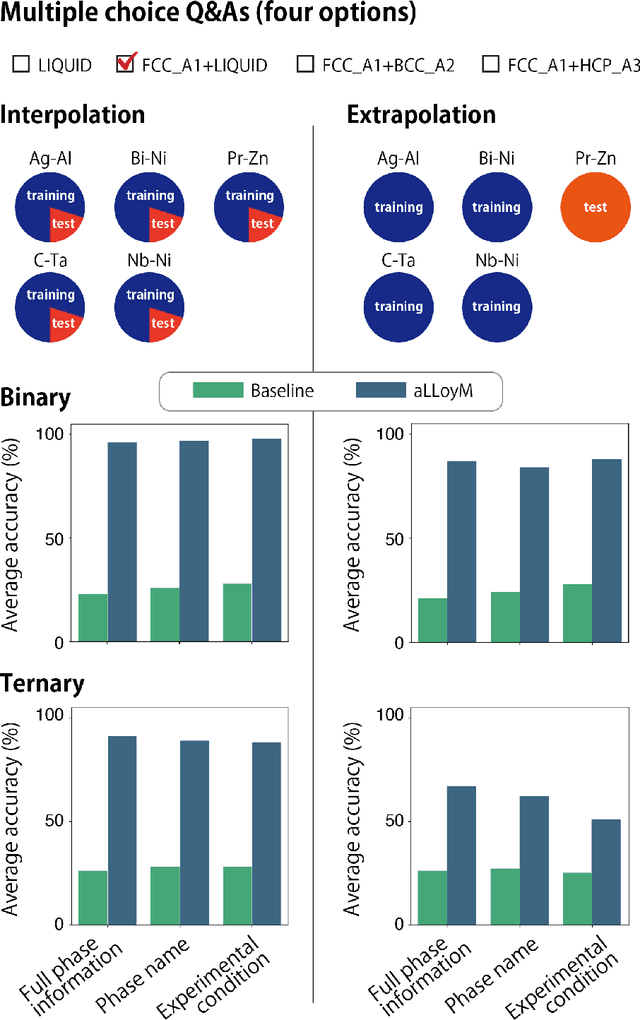
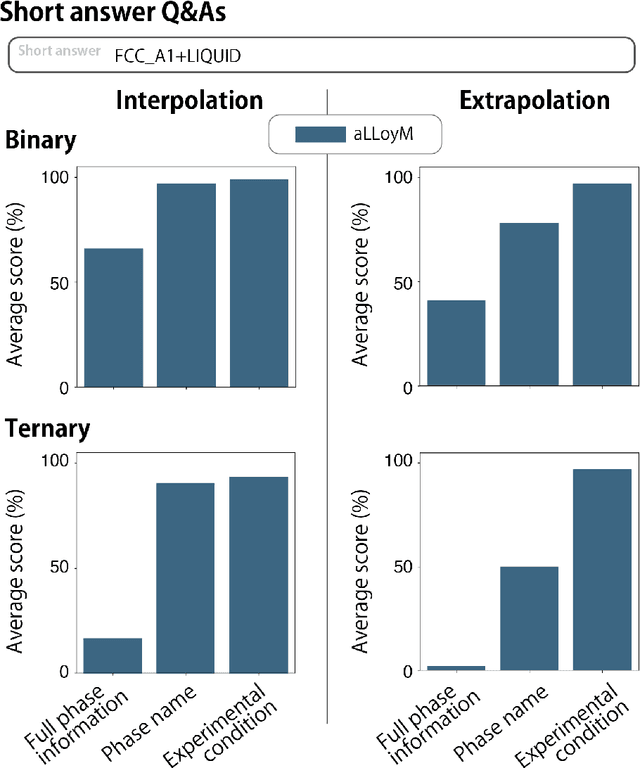
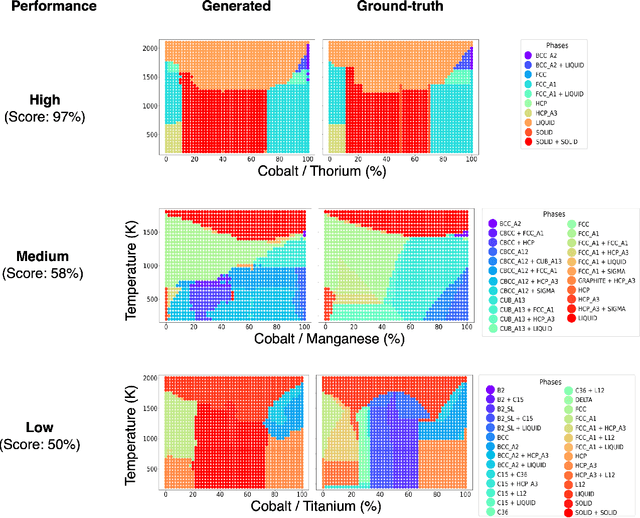
Large Language Models (LLMs) are general-purpose tools with wide-ranging applications, including in materials science. In this work, we introduce aLLoyM, a fine-tuned LLM specifically trained on alloy compositions, temperatures, and their corresponding phase information. To develop aLLoyM, we curated question-and-answer (Q&A) pairs for binary and ternary phase diagrams using the open-source Computational Phase Diagram Database (CPDDB) and assessments based on CALPHAD (CALculation of PHAse Diagrams). We fine-tuned Mistral, an open-source pre-trained LLM, for two distinct Q&A formats: multiple-choice and short-answer. Benchmark evaluations demonstrate that fine-tuning substantially enhances performance on multiple-choice phase diagram questions. Moreover, the short-answer model of aLLoyM exhibits the ability to generate novel phase diagrams from its components alone, underscoring its potential to accelerate the discovery of previously unexplored materials systems. To promote further research and adoption, we have publicly released the short-answer fine-tuned version of aLLoyM, along with the complete benchmarking Q&A dataset, on Hugging Face.
Inverse Design of Metamaterials with Manufacturing-Guiding Spectrum-to-Structure Conditional Diffusion Model
Jun 08, 2025Metamaterials are artificially engineered structures that manipulate electromagnetic waves, having optical properties absent in natural materials. Recently, machine learning for the inverse design of metamaterials has drawn attention. However, the highly nonlinear relationship between the metamaterial structures and optical behaviour, coupled with fabrication difficulties, poses challenges for using machine learning to design and manufacture complex metamaterials. Herein, we propose a general framework that implements customised spectrum-to-shape and size parameters to address one-to-many metamaterial inverse design problems using conditional diffusion models. Our method exhibits superior spectral prediction accuracy, generates a diverse range of patterns compared to other typical generative models, and offers valuable prior knowledge for manufacturing through the subsequent analysis of the diverse generated results, thereby facilitating the experimental fabrication of metamaterial designs. We demonstrate the efficacy of the proposed method by successfully designing and fabricating a free-form metamaterial with a tailored selective emission spectrum for thermal camouflage applications.
NbBench: Benchmarking Language Models for Comprehensive Nanobody Tasks
May 04, 2025Nanobodies, single-domain antibody fragments derived from camelid heavy-chain-only antibodies, exhibit unique advantages such as compact size, high stability, and strong binding affinity, making them valuable tools in therapeutics and diagnostics. While recent advances in pretrained protein and antibody language models (PPLMs and PALMs) have greatly enhanced biomolecular understanding, nanobody-specific modeling remains underexplored and lacks a unified benchmark. To address this gap, we introduce NbBench, the first comprehensive benchmark suite for nanobody representation learning. Spanning eight biologically meaningful tasks across nine curated datasets, NbBench encompasses structure annotation, binding prediction, and developability assessment. We systematically evaluate eleven representative models--including general-purpose protein LMs, antibody-specific LMs, and nanobody-specific LMs--in a frozen setting. Our analysis reveals that antibody language models excel in antigen-related tasks, while performance on regression tasks such as thermostability and affinity remains challenging across all models. Notably, no single model consistently outperforms others across all tasks. By standardizing datasets, task definitions, and evaluation protocols, NbBench offers a reproducible foundation for assessing and advancing nanobody modeling.
CRYSIM: Prediction of Symmetric Structures of Large Crystals with GPU-based Ising Machines
Apr 09, 2025Solving black-box optimization problems with Ising machines is increasingly common in materials science. However, their application to crystal structure prediction (CSP) is still ineffective due to symmetry agnostic encoding of atomic coordinates. We introduce CRYSIM, an algorithm that encodes the space group, the Wyckoff positions combination, and coordinates of independent atomic sites as separate variables. This encoding reduces the search space substantially by exploiting the symmetry in space groups. When CRYSIM is interfaced to Fixstars Amplify, a GPU-based Ising machine, its prediction performance was competitive with CALYPSO and Bayesian optimization for crystals containing more than 150 atoms in a unit cell. Although it is not realistic to interface CRYSIM to current small-scale quantum devices, it has the potential to become the standard CSP algorithm in the coming quantum age.
Preference-Optimized Pareto Set Learning for Blackbox Optimization
Aug 19, 2024



Multi-Objective Optimization (MOO) is an important problem in real-world applications. However, for a non-trivial problem, no single solution exists that can optimize all the objectives simultaneously. In a typical MOO problem, the goal is to find a set of optimum solutions (Pareto set) that trades off the preferences among objectives. Scalarization in MOO is a well-established method for finding a finite set approximation of the whole Pareto set (PS). However, in real-world experimental design scenarios, it's beneficial to obtain the whole PS for flexible exploration of the design space. Recently Pareto set learning (PSL) has been introduced to approximate the whole PS. PSL involves creating a manifold representing the Pareto front of a multi-objective optimization problem. A naive approach includes finding discrete points on the Pareto front through randomly generated preference vectors and connecting them by regression. However, this approach is computationally expensive and leads to a poor PS approximation. We propose to optimize the preference points to be distributed evenly on the Pareto front. Our formulation leads to a bilevel optimization problem that can be solved by e.g. differentiable cross-entropy methods. We demonstrated the efficacy of our method for complex and difficult black-box MOO problems using both synthetic and real-world benchmark data.
Molecule Graph Networks with Many-body Equivariant Interactions
Jun 19, 2024Message passing neural networks have demonstrated significant efficacy in predicting molecular interactions. Introducing equivariant vectorial representations augments expressivity by capturing geometric data symmetries, thereby improving model accuracy. However, two-body bond vectors in opposition may cancel each other out during message passing, leading to the loss of directional information on their shared node. In this study, we develop Equivariant N-body Interaction Networks (ENINet) that explicitly integrates equivariant many-body interactions to preserve directional information in the message passing scheme. Experiments indicate that integrating many-body equivariant representations enhances prediction accuracy across diverse scalar and tensorial quantum chemical properties. Ablation studies show an average performance improvement of 7.9% across 11 out of 12 properties in QM9, 27.9% in forces in MD17, and 11.3% in polarizabilities (CCSD) in QM7b.
Feature Importance Measurement based on Decision Tree Sampling
Jul 25, 2023Random forest is effective for prediction tasks but the randomness of tree generation hinders interpretability in feature importance analysis. To address this, we proposed DT-Sampler, a SAT-based method for measuring feature importance in tree-based model. Our method has fewer parameters than random forest and provides higher interpretability and stability for the analysis in real-world problems. An implementation of DT-Sampler is available at https://github.com/tsudalab/DT-sampler.
Efficient Model Selection for Predictive Pattern Mining Model by Safe Pattern Pruning
Jun 23, 2023



Predictive pattern mining is an approach used to construct prediction models when the input is represented by structured data, such as sets, graphs, and sequences. The main idea behind predictive pattern mining is to build a prediction model by considering substructures, such as subsets, subgraphs, and subsequences (referred to as patterns), present in the structured data as features of the model. The primary challenge in predictive pattern mining lies in the exponential growth of the number of patterns with the complexity of the structured data. In this study, we propose the Safe Pattern Pruning (SPP) method to address the explosion of pattern numbers in predictive pattern mining. We also discuss how it can be effectively employed throughout the entire model building process in practical data analysis. To demonstrate the effectiveness of the proposed method, we conduct numerical experiments on regression and classification problems involving sets, graphs, and sequences.
NIMS-OS: An automation software to implement a closed loop between artificial intelligence and robotic experiments in materials science
Apr 27, 2023NIMS-OS (NIMS Orchestration System) is a Python library created to realize a closed loop of robotic experiments and artificial intelligence (AI) without human intervention for automated materials exploration. It uses various combinations of modules to operate autonomously. Each module acts as an AI for materials exploration or a controller for a robotic experiments. As AI techniques, Bayesian optimization (PHYSBO), boundless objective-free exploration (BLOX), phase diagram construction (PDC), and random exploration (RE) methods can be used. Moreover, a system called NIMS automated robotic electrochemical experiments (NAREE) is available as a set of robotic experimental equipment. Visualization tools for the results are also included, which allows users to check the optimization results in real time. Newly created modules for AI and robotic experiments can be added easily to extend the functionality of the system. In addition, we developed a GUI application to control NIMS-OS.To demonstrate the operation of NIMS-OS, we consider an automated exploration for new electrolytes. NIMS-OS is available at https://github.com/nimsos-dev/nimsos.
On a linear fused Gromov-Wasserstein distance for graph structured data
Mar 09, 2022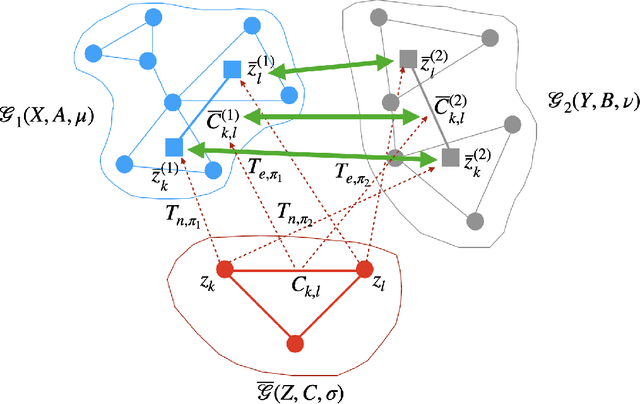
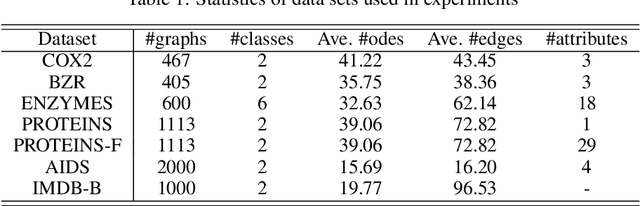
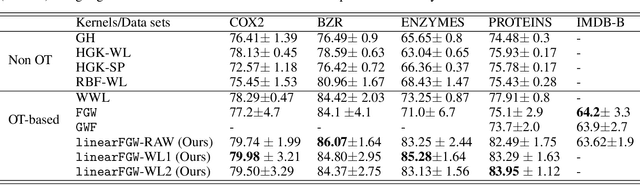
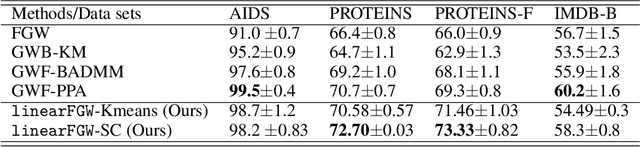
We present a framework for embedding graph structured data into a vector space, taking into account node features and topology of a graph into the optimal transport (OT) problem. Then we propose a novel distance between two graphs, named linearFGW, defined as the Euclidean distance between their embeddings. The advantages of the proposed distance are twofold: 1) it can take into account node feature and structure of graphs for measuring the similarity between graphs in a kernel-based framework, 2) it can be much faster for computing kernel matrix than pairwise OT-based distances, particularly fused Gromov-Wasserstein, making it possible to deal with large-scale data sets. After discussing theoretical properties of linearFGW, we demonstrate experimental results on classification and clustering tasks, showing the effectiveness of the proposed linearFGW.