 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInstruction-Tuned LLMs for Parsing and Mining Unstructured Logs on Leadership HPC Systems
Apr 06, 2026Leadership-class HPC systems generate massive volumes of heterogeneous, largely unstructured system logs. Because these logs originate from diverse software, hardware, and runtime layers, they exhibit inconsistent formats, making structure extraction and pattern discovery extremely challenging. Therefore, robust log parsing and mining is critical to transform this raw telemetry into actionable insights that reveal operational patterns, diagnose anomalies, and enable reliable, efficient, and scalable system analysis. Recent advances in large language models (LLMs) offer a promising new direction for automated log understanding in leadership-class HPC environments. To capitalize on this opportunity, we present a domain-adapted, instruction-following, LLM-driven framework that leverages chain-of-thought (CoT) reasoning to parse and structure HPC logs with high fidelity. Our approach combines domain-specific log-template data with instruction-tuned examples to fine-tune an 8B-parameter LLaMA model tailored for HPC log analysis. We develop a hybrid fine-tuning methodology that adapts a general-purpose LLM to domain-specific log data, enabling privacy-preserving, locally deployable, fast, and energy-efficient log-mining approach. We conduct experiments on a diverse set of log datasets from the LogHub repository. The evaluation confirms that our approach achieves parsing accuracy on par with significantly larger models, such as LLaMA 70B and Anthropic's Claude. We further validate the practical utility of our fine-tuned LLM model by parsing over 600 million production logs from the Frontier supercomputer over a four-week window, uncovering critical patterns in temporal dynamics, node-level anomalies, and workload-error log correlations.
Intelligent Sampling of Extreme-Scale Turbulence Datasets for Accurate and Efficient Spatiotemporal Model Training
Aug 05, 2025With the end of Moore's law and Dennard scaling, efficient training increasingly requires rethinking data volume. Can we train better models with significantly less data via intelligent subsampling? To explore this, we develop SICKLE, a sparse intelligent curation framework for efficient learning, featuring a novel maximum entropy (MaxEnt) sampling approach, scalable training, and energy benchmarking. We compare MaxEnt with random and phase-space sampling on large direct numerical simulation (DNS) datasets of turbulence. Evaluating SICKLE at scale on Frontier, we show that subsampling as a preprocessing step can improve model accuracy and substantially lower energy consumption, with reductions of up to 38x observed in certain cases.
Scaling Laws of Graph Neural Networks for Atomistic Materials Modeling
Apr 10, 2025Atomistic materials modeling is a critical task with wide-ranging applications, from drug discovery to materials science, where accurate predictions of the target material property can lead to significant advancements in scientific discovery. Graph Neural Networks (GNNs) represent the state-of-the-art approach for modeling atomistic material data thanks to their capacity to capture complex relational structures. While machine learning performance has historically improved with larger models and datasets, GNNs for atomistic materials modeling remain relatively small compared to large language models (LLMs), which leverage billions of parameters and terabyte-scale datasets to achieve remarkable performance in their respective domains. To address this gap, we explore the scaling limits of GNNs for atomistic materials modeling by developing a foundational model with billions of parameters, trained on extensive datasets in terabyte-scale. Our approach incorporates techniques from LLM libraries to efficiently manage large-scale data and models, enabling both effective training and deployment of these large-scale GNN models. This work addresses three fundamental questions in scaling GNNs: the potential for scaling GNN model architectures, the effect of dataset size on model accuracy, and the applicability of LLM-inspired techniques to GNN architectures. Specifically, the outcomes of this study include (1) insights into the scaling laws for GNNs, highlighting the relationship between model size, dataset volume, and accuracy, (2) a foundational GNN model optimized for atomistic materials modeling, and (3) a GNN codebase enhanced with advanced LLM-based training techniques. Our findings lay the groundwork for large-scale GNNs with billions of parameters and terabyte-scale datasets, establishing a scalable pathway for future advancements in atomistic materials modeling.
How does ion temperature gradient turbulence depend on magnetic geometry? Insights from data and machine learning
Feb 17, 2025Magnetic geometry has a significant effect on the level of turbulent transport in fusion plasmas. Here, we model and analyze this dependence using multiple machine learning methods and a dataset of > 200,000 nonlinear simulations of ion-temperature-gradient turbulence in diverse non-axisymmetric geometries. The dataset is generated using a large collection of both optimized and randomly generated stellarator equilibria. At fixed gradients, the turbulent heat flux varies between geometries by several orders of magnitude. Trends are apparent among the configurations with particularly high or low heat flux. Regression and classification techniques from machine learning are then applied to extract patterns in the dataset. Due to a symmetry of the gyrokinetic equation, the heat flux and regressions thereof should be invariant to translations of the raw features in the parallel coordinate, similar to translation invariance in computer vision applications. Multiple regression models including convolutional neural networks (CNNs) and decision trees can achieve reasonable predictive power for the heat flux in held-out test configurations, with highest accuracy for the CNNs. Using Spearman correlation, sequential feature selection, and Shapley values to measure feature importance, it is consistently found that the most important geometric lever on the heat flux is the flux surface compression in regions of bad curvature. The second most important feature relates to the magnitude of geodesic curvature. These two features align remarkably with surrogates that have been proposed based on theory, while the methods here allow a natural extension to more features for increased accuracy. The dataset, released with this publication, may also be used to test other proposed surrogates, and we find many previously published proxies do correlate well with both the heat flux and stability boundary.
Scalable Training of Graph Foundation Models for Atomistic Materials Modeling: A Case Study with HydraGNN
Jun 12, 2024We present our work on developing and training scalable graph foundation models (GFM) using HydraGNN, a multi-headed graph convolutional neural network architecture. HydraGNN expands the boundaries of graph neural network (GNN) in both training scale and data diversity. It abstracts over message passing algorithms, allowing both reproduction of and comparison across algorithmic innovations that define convolution in GNNs. This work discusses a series of optimizations that have allowed scaling up the GFM training to tens of thousands of GPUs on datasets that consist of hundreds of millions of graphs. Our GFMs use multi-task learning (MTL) to simultaneously learn graph-level and node-level properties of atomistic structures, such as the total energy and atomic forces. Using over 150 million atomistic structures for training, we illustrate the performance of our approach along with the lessons learned on two United States Department of Energy (US-DOE) supercomputers, namely the Perlmutter petascale system at the National Energy Research Scientific Computing Center and the Frontier exascale system at Oak Ridge National Laboratory. The HydraGNN architecture enables the GFM to achieve near-linear strong scaling performance using more than 2,000 GPUs on Perlmutter and 16,000 GPUs on Frontier. Hyperparameter optimization (HPO) was performed on over 64,000 GPUs on Frontier to select GFM architectures with high accuracy. Early stopping was applied on each GFM architecture for energy awareness in performing such an extreme-scale task. The training of an ensemble of highest-ranked GFM architectures continued until convergence to establish uncertainty quantification (UQ) capabilities with ensemble learning. Our contribution opens the door for rapidly developing, training, and deploying GFMs using large-scale computational resources to enable AI-accelerated materials discovery and design.
Data Distillation for Neural Network Potentials toward Foundational Dataset
Nov 09, 2023



Machine learning (ML) techniques and atomistic modeling have rapidly transformed materials design and discovery. Specifically, generative models can swiftly propose promising materials for targeted applications. However, the predicted properties of materials through the generative models often do not match with calculated properties through ab initio calculations. This discrepancy can arise because the generated coordinates are not fully relaxed, whereas the many properties are derived from relaxed structures. Neural network-based potentials (NNPs) can expedite the process by providing relaxed structures from the initially generated ones. Nevertheless, acquiring data to train NNPs for this purpose can be extremely challenging as it needs to encompass previously unknown structures. This study utilized extended ensemble molecular dynamics (MD) to secure a broad range of liquid- and solid-phase configurations in one of the metallic systems, nickel. Then, we could significantly reduce them through active learning without losing much accuracy. We found that the NNP trained from the distilled data could predict different energy-minimized closed-pack crystal structures even though those structures were not explicitly part of the initial data. Furthermore, the data can be translated to other metallic systems (aluminum and niobium), without repeating the sampling and distillation processes. Our approach to data acquisition and distillation has demonstrated the potential to expedite NNP development and enhance materials design and discovery by integrating generative models.
Scalable training of graph convolutional neural networks for fast and accurate predictions of HOMO-LUMO gap in molecules
Jul 22, 2022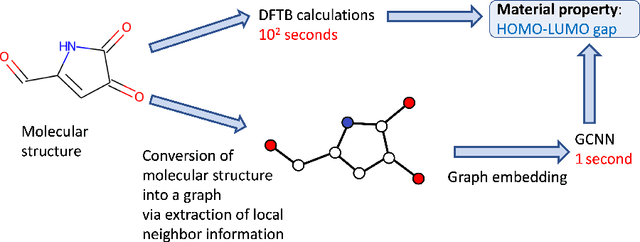

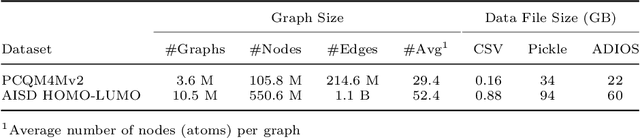
Graph Convolutional Neural Network (GCNN) is a popular class of deep learning (DL) models in material science to predict material properties from the graph representation of molecular structures. Training an accurate and comprehensive GCNN surrogate for molecular design requires large-scale graph datasets and is usually a time-consuming process. Recent advances in GPUs and distributed computing open a path to reduce the computational cost for GCNN training effectively. However, efficient utilization of high performance computing (HPC) resources for training requires simultaneously optimizing large-scale data management and scalable stochastic batched optimization techniques. In this work, we focus on building GCNN models on HPC systems to predict material properties of millions of molecules. We use HydraGNN, our in-house library for large-scale GCNN training, leveraging distributed data parallelism in PyTorch. We use ADIOS, a high-performance data management framework for efficient storage and reading of large molecular graph data. We perform parallel training on two open-source large-scale graph datasets to build a GCNN predictor for an important quantum property known as the HOMO-LUMO gap. We measure the scalability, accuracy, and convergence of our approach on two DOE supercomputers: the Summit supercomputer at the Oak Ridge Leadership Computing Facility (OLCF) and the Perlmutter system at the National Energy Research Scientific Computing Center (NERSC). We present our experimental results with HydraGNN showing i) reduction of data loading time up to 4.2 times compared with a conventional method and ii) linear scaling performance for training up to 1,024 GPUs on both Summit and Perlmutter.
Multi-task graph neural networks for simultaneous prediction of global and atomic properties in ferromagnetic systems
Feb 04, 2022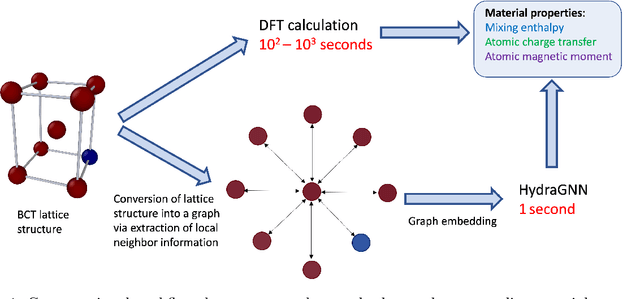

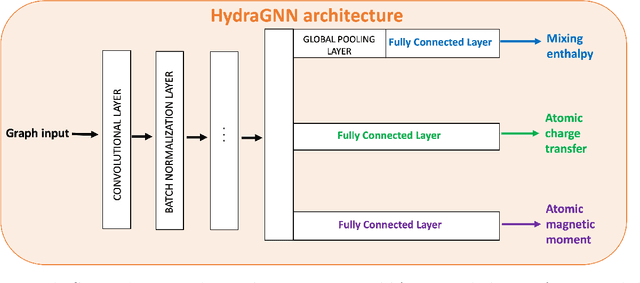
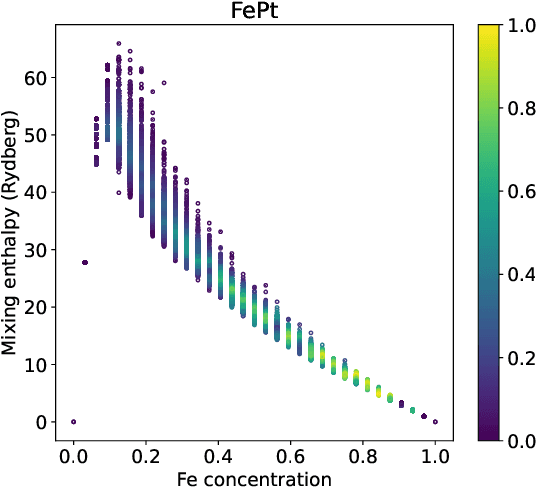
We introduce a multi-tasking graph convolutional neural network, HydraGNN, to simultaneously predict both global and atomic physical properties and demonstrate with ferromagnetic materials. We train HydraGNN on an open-source ab initio density functional theory (DFT) dataset for iron-platinum (FePt) with a fixed body centered tetragonal (BCT) lattice structure and fixed volume to simultaneously predict the mixing enthalpy (a global feature of the system), the atomic charge transfer, and the atomic magnetic moment across configurations that span the entire compositional range. By taking advantage of underlying physical correlations between material properties, multi-task learning (MTL) with HydraGNN provides effective training even with modest amounts of data. Moreover, this is achieved with just one architecture instead of three, as required by single-task learning (STL). The first convolutional layers of the HydraGNN architecture are shared by all learning tasks and extract features common to all material properties. The following layers discriminate the features of the different properties, the results of which are fed to the separate heads of the final layer to produce predictions. Numerical results show that HydraGNN effectively captures the relation between the configurational entropy and the material properties over the entire compositional range. Overall, the accuracy of simultaneous MTL predictions is comparable to the accuracy of the STL predictions. In addition, the computational cost of training HydraGNN for MTL is much lower than the original DFT calculations and also lower than training separate STL models for each property.